
Translate this page into:
Osmotic Demyelination Syndrome - Evolution of Extrapontine Before Pontine Myelinolysis on Magnetic Resonance Imaging
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Osmotic demyelination syndrome is a rare entity characterized by noninflammatory demyelination afflicting the central pons, basal ganglia, thalami, peripheral cortex, and hippocampi. Histopathologically, there is a destruction of myelin sheaths sparing the underlying neuronal axons due to the susceptibility of oligodendrocytes to rapid osmotic shifts often encountered in chronically debilitated patients. We present the temporal progression of signal abnormalities on sequential magnetic resonance imaging (MRI) scans in a middle-aged male initially presenting with altered mental status due to severe hyponatremia and subsequently developing rigidity, diagnosed as osmotic demyelination syndrome based on typical imaging findings and supportive laboratory parameters. This case demonstrates the importance of diffusion signal abnormalities as the first indicator of osmotic demyelination, evolution of extrapontine before pontine signal changes and the relative lack of correlation between patient's clinical profile and the extent of MRI signal abnormalities.
Keywords
Central pontine myelinolysis
diffusion-weighted magnetic resonance imaging
extrapontine myelinolysis
osmotic demyelination syndrome

INTRODUCTION
Osmotic demyelination syndrome is a debilitating, potentially fatal disorder which includes two components – central pontine myelinolysis (CPM) and extrapontine myelinolysis (EPM).[1] The CPM involves the basis pontis symmetrically, pontocerebellar fibers, and relatively spares the ventrolateral pons. EPM commonly involves the basal ganglia, thalami, and cerebral white matter.[1] The underlying physiology is theorized due to damage to oligodendrocytes secondary to rapid changes in osmolarity. Magnetic resonance imaging (MRI) is the preferred imaging modality for the diagnosis and subsequent follow-up of brain parenchymal abnormalities due to this disorder.
We present the spectrum of imaging findings in a case diagnosed with osmotic demyelination syndrome demonstrating the temporal progression of signal abnormalities on sequential MRI scans. This case establishes the sensitivity of diffusion-weighted imaging (DWI) in picking up early signal alterations as well as demonstrates the onset of EPM preceding pontine demyelination.
CASE REPORT
A middle aged, nondiabetic, and nonhypertensive male, from a well-to-do family, presented to the emergency with a feeling of generalized weakness of 1-week duration and acute-onset altered sensorium. Patient's vitals at presentation were stable. On initial examination, the patient was conscious, oriented in time, place, and person, appeared listless, mildly drowsy, and demonstrated delayed responses to verbal commands. No specific motor or sensory weakness or any focal neurological deficit was present.
The MRI scan done on a GE Explorer 1.5 Tesla silent MRI system at initial presentation (day 1) in the emergency department revealed no signal abnormality in the brain parenchyma. No acute infarct, intracranial hemorrhage, or any focal lesion was seen in Figures 1 and 2. The sequences taken in the routine MRI brain protocol included DWI with apparent diffusion coefficient (ADC) maps, T1-weighted and T2-weighted fast spin-echo images, fluid attenuation inversion recovery (FLAIR) images, and susceptibility-weighted gradient-echo sequences.
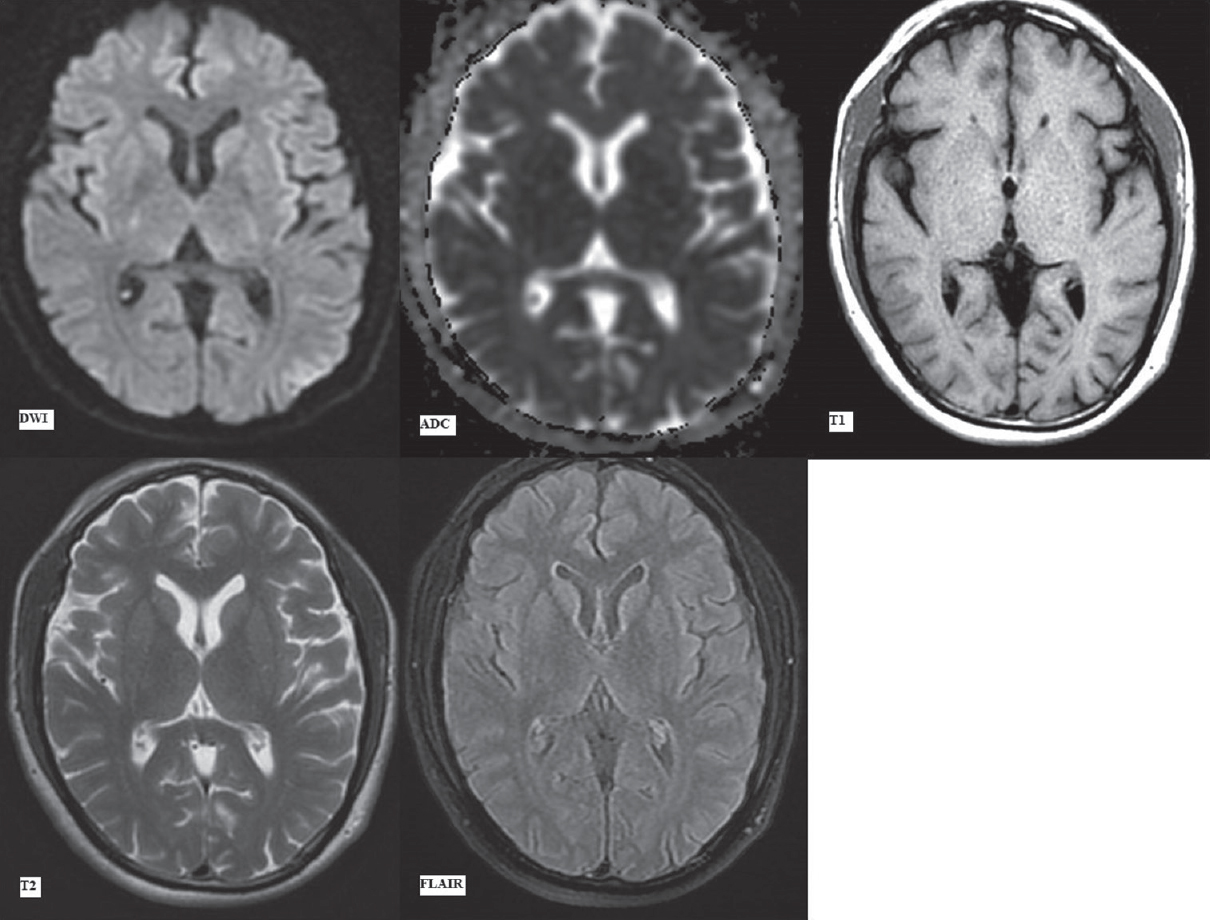
- First magnetic resonance imaging at presentation (day 1). Diffusion-weighted imaging with apparent diffusion coefficient map, T1-weighted, T2-weighted and fluid attenuation inversion recovery images (as labeled) at the level of pons showing no signal alteration

- First magnetic resonance imaging at presentation (day 1). Diffusion-weighted imaging, apparent diffusion coefficient map, T2-weighted and fluid attenuation inversion recovery images (as labelled) at the level of basal ganglia showing no signal alteration
Results of routine laboratory investigations at presentation revealed serum sodium (Na) level to be 102 mmol/L and serum potassium (K) level to be 3.1 mmol/L suggestive of severe hyponatremia (hyponatremia defined as sodium level <136 mmol/L, with severe hyponatremia defined as sodium level <120 mmol/L) with hypokalemia (corresponds to potassium level <3.5 mmol/L). This was presumed to be the cause of the initial metabolic encephalopathy. Ultrasound examination of the abdomen revealed that the kidneys and the liver were normal in appearance and subsequently confirmed by normal serum creatinine values and liver function tests. Possible etiology of the electrolyte imbalance was initially suspected to be malnutrition. The patient's initial workup, however, revealed no evidence of elevated blood or urine ketone bodies or any other laboratory parameter to prove malnutrition which was further confirmed on subsequent detailed history as the patient's neurological status improved. The patient later divulged that he did drink alcohol occasionally (approximately 40–50 mL of alcohol up to a maximum of 2 times a week), but the quantities and frequency are too low to consider him as a chronic alcoholic. This electrolyte imbalance was the first such episode for the patient and its cause, therefore, remained elusive.
The patient was shifted to the intensive care unit (ICU), and correction of the osmotic derangements was commenced. He was started on intravenous hypertonic saline 3% in ICU, administered slowly with monitoring of the serum electrolytes, twice daily. The next day after initiation of correction, the serum sodium had increased to 109 mmol/L (still severe hyponatremia but with sudden significant jump from the previous value) with an elevated serum osmolality of 314 mosm/kg. There was no change in the treatment regimen, and the abrupt initial fluctuations occurred in spite of utmost care to administer hypertonic saline slowly according to clinical guidelines. The patient did not develop hypernatremia at any point during correction.
In the next 24 h, the serum sodium increased to 120.7 mmol/L [Table 1]. Later, an average correction of 4–8 mmol/day was attained to reach a normal value of 140.2 mmol/L on day 11. In the interim, the initial low potassium also fluctuated, eventually normalizing by day 14 after admission.

During the course of correction, the patient's encephalopathy improved initially, and the patient was shifted to ward. However, on day 7, the patient's clinical examination revealed the onset of rigidity (extrapyramidal tract symptoms) with worsening drowsiness which was not present earlier. This finding was disconcerting as the electrolyte status of the patient was on corrective course, almost normalized, and hence warranting a repeat MRI examination to ascertain the cause of this new sign. He was shifted to ICU again.
MRI brain performed at this juncture revealed areas of symmetrical restricted diffusion (hyperintense signal on diffusion B-1000 images with the corresponding hypointense signal on ADC maps) in bilateral caudate nuclei and putamen with a small focus of restricted diffusion in the central pons [Figures 3 and 4].

- Magnetic resonance imaging brain findings on day 7. Diffusion-weighted imaging, apparent diffusion coefficient map, susceptibility-weighted imaging, fluid attenuation inversion recovery, T2-weighted and T1-weighted images (as labeled) at the level of pons. Arrows showing focal hyperintensity in the central pons on diffusion-weighted imaging with corresponding signal drop on apparent diffusion coefficient suggestive of restricted diffusion. susceptibility-weighted imaging, fluid attenuation inversion recovery, T2-weighted and T1-weighted images show no signal alteration

- Magnetic resonance imaging brain findings on day 7. Diffusion-weighted imaging, apparent diffusion coefficient map, T1-weighted, T2-weighted, fluid attenuation inversion recovery and susceptibility-weighted imaging images (as labeled) at the level of basal ganglia. Arrows showing hyperintense signal on diffusion-weighted imaging and subsequent apparent diffusion coefficient image showing corresponding signal drop in bilateral caudate nuclei (upper arrows) and putamina (lower arrows), suggestive of restricted diffusion. Hyperintense signal in these areas (upper and lower arrows) on T2-weighted image and fluid attenuation inversion recovery image. No signal alteration on T1-weighted image and susceptibility-weighted imaging image
In addition, bilateral caudate nuclei and putamen were symmetrically hyperintense on FLAIR/T2-weighted images with no signal alteration on T1-weighted images [Figure 4]. The focal, central pontine diffusion restriction, on the other hand, showed no signal abnormality on other sequences [Figure 4]. This implied that the involvement of these areas was of recent onset. In addition, the basal ganglia signal abnormalities had already begun to evolve, being demonstrable on T2 and FLAIR images while the pontine signal abnormality which showed only diffusion restriction had developed subsequently, i.e., more acute in onset.
The patient continued to have waxing and waning course of extrapyramidal tract symptoms, mainly rigidity and abnormal plantar reflexes. Lumbar puncture was performed for evaluation of cerebrospinal fluid (CSF) at this juncture to rule out any contributory infective or autoimmune etiology. The CSF parameters were all within normal limits. A follow-up MRI scan was then advised to evaluate for any progression, even though the clinical profile remained almost static.
Subsequently, 3rd MRI brain was performed after 12 days of the 2nd MRI (day 19 since admission). Clinically, the patient had fluctuating mild rigidity as described earlier. The MRI changes had however progressed.
Bilateral caudate nuclei and putamina, previously showing restricted diffusion, now exhibited hyperintense signal on DWI without any corresponding signal drop on ADC. This is likely due to T2 shine through effect with normalization of ADC signal with time. These areas remained symmetrically hyperintense on FLAIR/T2 images with hypointense signal on T1-weighted images [Figure 5]. The involved areas also appear to be swollen on these images.

- Magnetic resonance imaging brain findings on day 19. Diffusion-weighted imaging, apparent diffusion coefficient, T1-weighted, T2-weighted, fluid attenuation inversion recovery and susceptibility-weighted imaging images (as labeled) at the level of basal ganglia. Hyperintense signal on diffusion-weighted imaging in bilateral caudate nuclei (upper arrows) and putamina (lower arrows) without any signal drop on apparent diffusion coefficient. Subsequent images show symmetrical hypointense signal on T1-weighted and hyperintense signal on T2 and fluid attenuation inversion recovery images in bilateral caudate nuclei (upper arrows) and putamina (lower arrows)
The small pontine focus of restricted diffusion had increased in extent to involve the entire central pontine tegmentum and basis pontis sparing the ventrolateral pons (and possibly sparing the corticospinal tracts situated anteriorly). It showed a trident-shaped signal abnormality on DWI/ADC maps. This included central hyperintensity surrounded by a thin rim of less hyperintense signal on DWI. On ADC, the central pons demonstrated mildly hypointense signal with peripheral mildly hyperintense rim (suggestive of central cytotoxic edema surrounded by a rim of vasogenic edema). These areas showed uniform hyperintense signal on FLAIR/T2-weighted images and hypointense signal on T1-weighted images [Figure 6]. The pattern of these signal abnormalities is highly suggestive of CPM with extrapontine involvement in the basal ganglia.

- Magnetic resonance imaging findings on day 19. Diffusion-weighted imaging, apparent diffusion coefficient map, T1-weighted, T2-weighted, fluid attenuation inversion recovery and susceptibility-weighted imaging images (as labeled) at the level of pons. Arrows showing trident shaped pontine signal abnormality on diffusion-weighted imaging and apparent diffusion coefficient images with central restricted diffusion (hyperintense on diffusion-weighted imaging, hypointense on apparent diffusion coefficient) surrounded by rim of vasogenic edema (minimally hyperintense on diffusion-weighted imaging, thin hyperintense rim on apparent diffusion coefficient due to intrinsic T2 effect). Subsequent images with arrows depicting hypointense T1-weighted and hyperintense T2/fluid attenuation inversion recovery signal alteration in central pons. No abnormality is seen on susceptibility-weighted imaging images
Subsequently, MRI brain was performed after 17 days of the previous MRI (day 36 since admission), before patient's discharge. Clinically, patient's drowsiness and altered mental status had resolved. Mild residual extrapyramidal tract rigidity remained which had also shown significant interval recovery. This MRI revealed that the central pontine involvement remained almost unchanged, although marginally smaller on FLAIR/T2 images due to reduction in the vasogenic edema [Figures 7 and 8]. Bilateral caudate and lentiform nuclei showed persistent hyperintense signal on T2 and FLAIR images [Figure 9], with reduction in swelling. The diffusion signal abnormalities had temporally evolved as expected. The patient, in spite of extensive signal abnormality in pons and bilateral basal ganglia, was well preserved with minimal rigidity being the only neurological sequelae.

- Magnetic resonance imaging findings on day 36. Diffusion-weighted imaging, apparent diffusion coefficient map, T1-weighted, T2-weighted, fluid attenuation inversion recovery and susceptibility-weighted imaging images (as labeled) at the level of pons. Arrows depict trident-shaped mild hyperintensity on diffusion-weighted imaging and apparent diffusion coefficient images, hyperintense signal on T2-weighted and fluid attenuation inversion recovery images with corresponding hypointensity on T1-weighted images in central pontine tegmentum and basis pontis sparing the ventrolateral pons. The extent of signal abnormalities has marginally decreased due to a reduction in vasogenic edema as compared to the previous magnetic resonance imaging

- Magnetic resonance imaging findings on day 36. T2-weighted sagittal and coronal sections (as labeled) at the level of pons. Arrows depict the extent of T2-weighted signal abnormality in central pons characteristically sparing the ventrolateral pons

- Magnetic resonance imaging findings on day 36. Diffusion-weighted imaging, apparent diffusion coefficient map, T1-weighted, T2-weighted, and fluid attenuation inversion recovery images (as labeled) at the level of basal ganglia. diffusion-weighted imaging image showing symmetrical hyperintense signal in bilateral caudate nuclei (upper arrows) and putamina (lower arrows) without any apparent diffusion coefficient reversal. Symmetrical corresponding T1 hypointense, T2 hyperintense, and fluid attenuation inversion recovery hyperintense signal in bilateral caudate nuclei (upper arrows) and putamina (lower arrows). There is a reduction in edema in these areas as compared to the previous magnetic resonance imaging
The patient was discharged in a stable condition, advised physiotherapy for residual rigidity and is on regular follow-up. The patient at last follow-up has no neurological sequelae, and the initial sign of rigidity has resolved suggestive of complete recovery.
DISCUSSION
Osmotic demyelination syndrome was first described in 1959 as a noninflammatory central pontine demyelination occurring in chronic alcoholic and malnourished patients.[2] It was later discovered that the process of osmotic demyelination is not only limited exclusively to the pons but also involves various extrapontine sites, a condition referred to as EPM.[1] The CPM involves the basis pontis symmetrically, pontocerebellar fibers, and relatively sparing the ventrolateral pons and corticospinal tracts. Sites of EPM include the basal ganglia, cerebral white matter, the peripheral cortex, hippocampi, and lateral geniculate bodies.[1] EPM generally occurs contemporaneously with CPM; however, case reports suggesting that it may precede CPM[3] or occur in isolation also exist.[4]
Pathophysiologically, severe hyponatremia, without a rise in other osmoles to balance extracellular osmolality, will cause water to enter across the cell membranes into the cells. In the brain cells, this will result in potentially lethal cerebral edema. There are several homeostatic mechanisms to maintain cell volume in this setting of serum hypotonicity, termed as “regulatory volume decrease.”[5] In the brain, the first corrective step precedes regulatory volume decrease and involves influx of interstitial fluid, rich in sodium, into the hypoosmolar CSF.[67] Subsequently, potassium is lost to compensate for the sodium loss. It was realized that additionally, organic solutes, such as myoinositol, taurine, and glutamate, contribute to regenerate equilibrium and are gradually lost over a period of several days to render the cell isotonic to the extracellular fluid, thereby achieving homeostasis.
With the process of sodium recorrection, the extracellular environment now suddenly becomes hypertonic to the intracellular medium. The intracellular influx of electrolytes in a hypertonic extracellular environment is, however, quite different as compared to their loss from the cell in a chronic hypoosmolar state due to hyponatremia. The initial homeostatic mechanisms will involve the movements of inorganic ions, which the cells will already be depleted in due to prior homeostatic measures. Hence, if they are exhausted without achieving equilibrium, the cells are now left with only a few options, i.e., either to synthesize organic osmoles or transport them from the extracellular environment – both of which cost time and cellular energy. However, if the rate of the rise of extracellular tonicity is still in excess, as might happen with rapid recorrection, the cell will be unable to maintain equilibrium and will shrink. The oligodendrocytes are especially vulnerable to death from such a volume loss. It is speculated that this intracellular synthesis and transport of organic osmoles at a rapid rate to counter hypertonicity might be related to the patient's nutritional status.[5] It is, however, impossible to calculate a threshold value for the rate of change of osmolality that may be considered as safe.[8] It is also well established that progressive cellular shrinkage induced by hypertonicity triggers apoptosis and leads to eventual cell death.
This leads to the conclusion that sodium correction should be slow in such cases. However, if it is too slow, the patient might not recover from the hyponatremia-induced cerebral edema. In addition, the rapidity of initial rise in the serum sodium concentration, even while avoiding hypertonic saline, or while carefully supervising a slow increase, is quite unpredictable and may overshoot in spite of utmost precautions.[5]
On histopathology, CPM is characterized by intramyelinitic splitting, vacuolization, and rupture of myelin sheaths with preserved axons.[9]
Several initial case studies elucidated the conditions precipitating this entity. It was determined that it was frequently related to rapid correction of hyponatremia (>10 mmol/L/day).[1011] It also occurred with gradual correction of hyponatremia as well as in normonatremic and hypernatremic patients.[12131415] Several conditions predisposing patients to its development were also found. These include renal failure patients on dialysis, liver failure, and transplantation, malignancies such as lymphoma, carcinoma, cachexia, sepsis, acute pancreatitis, pellagra, and chronic alcoholism.[1617] Chronic alcoholism-associated CPM tends to be benign with a favorable prognosis compared to CPM secondary to rapid correction of hyponatremia.[1819] Furthermore, CPM has been documented to occur in hypokalemic patients even with a steady rise in serum sodium levels.[2021]
In our case, no predisposing factor was present. His liver and kidney function tests were within normal range. The patient did drink alcohol occasionally (approximately 40–50 mL of alcohol up to a maximum of 2 times a week) but not frequently enough to consider him as a chronic alcoholic.
The clinical course of CPM is biphasic,[16] beginning with an initial phase of encephalopathy or seizures caused by the hyponatremia, which improves with correction of osmotic imbalance. This is followed by neurological deterioration, usually occurring a few days after the onset of correction, incited by the osmotic myelinolysis. This latter phase manifests as quadriparesis and pseudobulbar palsy (due to the involvement of corticospinal and corticobulbar tracts in pons). Extrapyramidal symptoms, such as rigidity, myoclonus, movement disorders, and parkinsonism-like features in patients with EPM, have also been reported. Once osmotic demyelination sets in it is usually only partially reversible has a protracted course with high morbidity and potential mortality, thus prevention and early detection are most important.
In our case too, the clinical course was biphasic with initial mild metabolic encephalopathy, followed by a short interval of improvement and later onset of worsening sensorium and extrapyramidal tract symptoms. Thereafter, in spite of extensive pontine and basal ganglia involvement on MRI, the patient did not have significant symptoms, and extrapyramidal tract sign rigidity was the only finding.
Usually, the clinical findings lag behind the typical imaging findings by about 1–2 weeks as has been reported in previous studies. MRI is more sensitive than computed tomography in picking up the changes. The earliest and the only reported abnormality that may be picked up initially is diffusion restriction within 24 h of onset of clinical symptoms.[22] The diffusion restriction usually settles by 3 weeks.[23]
This case similarly reaffirms the importance of DWI in diagnosis. The diffusion restriction in basal ganglia corresponded to the worsening of symptoms in our case and was seen within 7 days of starting the correction. More importantly, focal restricted diffusion was the only initial pontine abnormality, and the patient's clinical profile warranted a high degree of suspicion for osmotic demyelination. This was confirmed on subsequent imaging. The corresponding initial ADC signal drops in basal ganglia and pons normalized over the course of subsequent 2–3 weeks. The FLAIR/T2-hyperintense signal appeared after diffusion restriction, and the T1-weighted signal alteration was the last to appear. The altered signal on FLAIR/T2-weighted imaging persisted until discharge.
Classical CPM shows hyperintensity in the central pons on T2-weighted and FLAIR images. This may evolve into a hyperintense “trident-shaped” central pontine signal, with relative sparing of the ventrolateral pons.[242526] The extrapontine involvement is also similar in progression with diffusion restriction and abnormal signal on T2-weighted/FLAIR images. In this case, the extrapontine basal ganglia involvement was florid at 7 days while pontine involvement was minimal at outset, seen only as a small focus of restricted diffusion. Over the course of the next 12 days, central pontine involvement progressed to the typical trident pattern, thereby clinching the diagnosis.
In conclusion, the implications are as follows. First, it proves that diffusion restriction is the first signal abnormality to appear. Second, the caudate nucleus and putamen signal abnormalities preceded the pontine signal abnormalities, i.e., EPM preceded, possibly by a few hours, the CPM. Third, the territorial involvement, although characteristic once the imaging findings have evolved, is initially nonspecific especially if extrapontine precedes pontine involvement and needs a high index of clinical and radiological suspicion for accurate diagnosis.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- Central pontine myelinolysis: A hitherto undescribed disease occurring in alcoholic and malnourished patients. AMA Arch Neurol Psychiatry. 1959;81:154-72.
- [Google Scholar]
- Osmotic myelinolysis: Does extrapontine myelinolysis precede central pontine myelinolysis? Report of two cases and review of literature. Indian J Radiol Imaging. 2015;25:177-83.
- [Google Scholar]
- Central pontine and extrapontine myelinolysis: The osmotic demyelination syndromes. J Neurol Neurosurg Psychiatry. 2004;75(Suppl 3):iii22-8.
- [Google Scholar]
- Treating hyponatremia: Damned if we do and damned if we don’t. Kidney Int. 1990;37:1006-18.
- [Google Scholar]
- Central pontine and extrapontine myelinolysis after correction of hyponatraemia. The Neurologist. 2000;6:255-66.
- [Google Scholar]
- Graham DI, Lantos PL, eds. Greenfield's Neuropathology (7th ed). London: Arnold/Oxford University Press; 2002.
- Rapid correction of hyponatremia causes demyelination: Relation to central pontine myelinolysis. Science. 1981;211:1068-70.
- [Google Scholar]
- Association between rise in serum sodium and central pontine myelinolysis. Ann Neurol. 1982;11:128-35.
- [Google Scholar]
- Cerebral encephalopathy with extrapontine myelinolysis in a case of postpartum hypernatremia. Indian J Radiol Imaging. 2014;24:57-60.
- [Google Scholar]
- Osmotic demyelination syndrome in a normonatremic patient of chronic kidney disease. Indian J Crit Care Med. 2014;18:609-11.
- [Google Scholar]
- Case report: MRI demonstration of pontine and thalamic myelinolysis in a normonatremic alcoholic. Clin Radiol. 1993;47:137-8.
- [Google Scholar]
- Severe hyponatraemia secondary to beer potomania complicated by central pontine myelinolysis. Int J Clin Pract. 1998;52:585-7.
- [Google Scholar]
- A review of the causes of central pontine myelinosis: Yet another apoptotic illness? Eur J Neurol. 2001;8:103-9.
- [Google Scholar]
- Benign type of central pontine myelinolysis in alcoholism – clinical, neuroradiological and electrophysiological findings. J Neurol. 2003;250:1077-83.
- [Google Scholar]
- Central pontine myelinolysis in a chronic alcoholic: A clinical and brain magnetic resonance imaging follow-up. Vojnosanit Pregl. 2013;70:785-8.
- [Google Scholar]
- Osmotic demyelination syndrome following correction of hyponatremia: Association with hypokalemia. Am J Med. 1994;96:408-13.
- [Google Scholar]
- Centropontine myelinolysis after correction of hyponatremia: Role of associated hypokalemia. Clin Nephrol. 2007;67:345-51.
- [Google Scholar]
- Early diagnosis of central pontine myelinolysis with diffusion-weighted imaging. AJNR Am J Neuroradiol. 2004;25:210-3.
- [Google Scholar]
- Decreased diffusion in central pontine myelinolysis. AJNR Am J Neuroradiol. 2001;22:1476-9.
- [Google Scholar]
- Temporal changes of MR findings in central pontine myelinolysis. AJNR Am J Neuroradiol. 1995;16:975-7.
- [Google Scholar]
- Outcome of central pontine and extrapontine myelinolysis (n=44) J Neurol. 1999;246:700-5.
- [Google Scholar]
- Central pontine myelinolysis and its imitators: MR findings. Radiology. 1988;168:795-802.
- [Google Scholar]




