
Translate this page into:
Multiple, Primary Brain Tumors with Diverse Origins and Different Localizations: Case Series and Review of the Literature
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background:
Multiple, primary brain tumors with different histological types occurring in the same patient are extremely rare. Several hypotheses have been proposed, and the pathophysiology of coexisting tumors has long been debated; however, due to low incidence, standard practices for this scenario are still inconclusive.
Case Description:
The authors describe 6 cases of coexisting tumors. By conducting a literature research focused on the computed tomography (CT) era and patients without prior radiation or phakomatosis. Sixty-five such reported cases were identified. In addition, the authors summarize their experience in 6 patients including histopathological features, chronological presentations, outcomes, mortality, and management from their series as well as from previous cases from the reported literature.
Conclusion:
The coexistence of multiple, primary brain tumors is an interesting condition. Surgical management remains the major treatment; malignant histology has a poor prognostic factor.
Keywords
Coexistence of brain tumor
diffuse astrocytoma
glioblastoma
meningioma
multiple primary brain tumor

INTRODUCTION
The association of multiple, primary brain tumors with different histology occurring simultaneously in the same patient is an extremely rare condition. It has been frequently associated with previous radiotherapy or phakomatosis[12] but is still exceptionally rare as a primary observation. Before the era of computed tomographies (CTs), Cushing and Eisenhardt reported, in 1938, a patient who harbored both a glioblastoma and a meningioma.[3] Only a few cases of this scenario have since been reported. The authors present their experience gained from a series of such primary brain tumors with different histopathology types and in different intracranial localizations. They discuss these cases in the context of the currently available literature.
The most frequently reported primary brain tumor in the Central Brain Tumor Registry of the United States is nonmalignant meningioma (36.1%), followed by glioblastoma (15.4%). In addition, the nonmalignant pituitary adenoma and nerve sheath tumors account for 15.1% and 8% of all tumors, respectively. In gender-specific distribution, the incidence rate of tumor of the meninges is higher in females (10.51/100,000) rather than in males (4.85/100,000), while the incidence rate of the tumor of the neuroepithelial tissue is higher in males (7.77/100,000) rather than in females (5.61/100,000).[4] Moreover, the incidental, primary brain tumor can be found in general population. Asymptomatic meningiomas were the most frequent in 0.9%–1% while pituitary adenomas were 0.3% within the population.[56] However, the condition where coexisting, primary brain tumors occur in the same patient is truly scarce.
We reviewed these from the relevant literature in MEDLINE, EMBASE, and Scopus. The selection criteria included those in the English language and illustrated neuroimaging such as CT or magnetic resonance imaging (MRI) brain scans. The authors also do not consider these tumors to be associated with previous radiotherapy (Cahan's criteria) or phakomatosis.[7] According to the criteria, 65 cases were reported in the literature and the authors added 6 cases in Appendix 1.[89101112131415161718192021222324252627282930313233343536373839]
CASE SERIES
Patient 1: Meningioma and diffuse astrocytoma
A 68-year-old, right-handed women suffering from dementia on the right arm coupled with suffering leg weakness in October 2013. A clinical examination showed right hemiparesis and right facial nerve palsy with upper motor neuron type. She had neither café-au-lait spots nor Lisch nodules, which are found in neurofibromatosis Type 1 (NF1). There was also an absence of first-degree relatives of NF1.
MRI of the brain showed a homogeneously enhancing mass with dural tail sign located in the left frontal lobe as seen on T1-weighted (T1W) gadolinium-enhanced images. Furthermore, another lesion was observed at the right insular lobe that showed an ill-defined area of mixed hypo- and hyperintense but nonenhancing lesion on T1W, fluid-attenuated inversion recovery, and T1W gadolinium-enhanced images, respectively [Figure 1a]. In addition, the T1-weight inversion recovery MRI showed hyperintensity of the right insular lesion [Figure 1b].
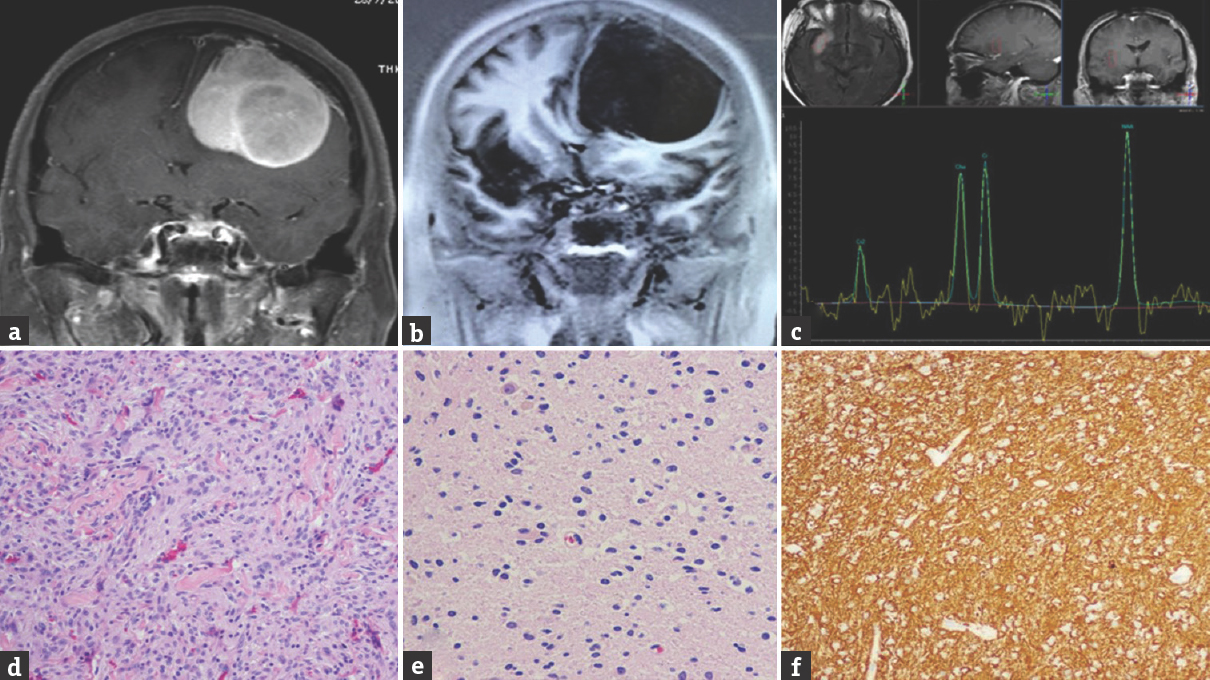
- Case 1 – Magnetic resonance imaging showing an enhanced mass with a dural tail sign at the left frontal convexity (a). T1-weight inversion recovery magnetic resonance imaging showing hyperintense lesion at the right insular lobe (b). Magnetic resonance spectroscopy showing decreased N-acetylaspartate without elevated choline at right temporal lesion (c). Histological findings of a fibrous meningioma demonstrating a proliferation of generally spindled cells (d). Histological findings of diffused astrocytoma demonstrating increased cellular density of glial cell without necrosis (e). Immunoreactivity of astrocytoma was positive for glial fibrillary acidic protein (f)
A left craniotomy for tumor removal was performed to address the frontal lesion first. The tumor was found attached to the dura mater and measuring 4.5 cm in diameter by 4.5 cm. Both tumor and attached dura mater were completely removed. After surgery, this patient made an uneventful recovery without any new neurological impairment. The pathology of the frontal lesion was fibrous meningioma (WHO II) [Figure 1d].
One year after the first surgery, the patient suffered a generalized tonic–clonic seizure. A follow-up MRI brain scan showed no evidence of recurrence of the left frontal meningioma, and there was a stable size of the right insular lesion. In addition, no decreased N-acetylaspartate (NAA) and no choline peak were noted in magnetic resonance spectroscopy [Figure 1c]. Taking the clinical seizure as a sign of progression, we had the indications that an operation was required to obtain a diagnosis. A right-sided craniotomy for a transsylvian approach was performed. Discoloration of the insular cortex was encountered, and the extent of resection was 90% in the complex location. Thus, surgical specimens were sent for analysis.
Pathological examination of the specimens found hypercellularity of glial cells without endothelial proliferation and necrosis. Furthermore, the specimen stained positive for glial fibrillary acidic protein (GFAP), which indicated a glial cell tumor. Due to the lack of diagnostic molecular testing, a final diagnosis of tumor was designated as those not otherwise specified diffuse astrocytoma (WHO II) according to the 2016 WHO classification [Figure 1e and f]. After surgery, this patient again made an uneventful recovery without any new neurological impairment. At follow-up, she maintained independent daily activities.
Chromosome analysis
Chromosomal analysis from peripheral blood cells of the patient was performed for screening germline mutation. The karyotype was 44, XX and an absence of chromosome 17p deletions.
Patient 2: Glioblastoma multiforme with pituitary adenoma
A 63-year-old male patient was referred to our hospital due to an alteration in his consciousness. Two weeks prior, he had suffered from a change in mentation and left hemiparesis, so the patient had been admitted to a provincial hospital. CT head scans demonstrated two sites with intracranial tumors: an irregular rim-enhancing mass at the right frontoparietal lobe and a sellar tumor. During hospitalization, the patient showed progressive neurological deterioration. Therefore, the patient underwent a decompressive craniectomy and was transferred to our hospital. At the tertiary hospital, he was fully oriented but had hemiplegia on the left side. Cranial T1W MRI with intravenous (I/V) gadolinium revealed an irregular, heterogeneously contrast-enhancing mass at the right frontoparietal lobe measuring 52 mm × 68 mm × 54 mm with central hypointensity indicative of necrosis [Figure 2a].

- Case 2 – Magnetic resonance imaging showing an enhanced mass with central necrosis at the right frontoparietal lobe (a). Palisading tumor necrosis was observed (b). The neoplastic glial cells have nuclear enlargement with hyperchromatic nuclei, irregular nuclear membrane, and abnormal endothelial cell proliferation (c). Magnetic resonance imaging showing enhanced suprasellar mass (d). Sagittal magnetic resonance imaging showing enhanced sellar-suprasellar mass compressed chiasm (e). Pituitary tumor showing proliferation of homogeneous round cell with sheeting pattern. The tumor cells have eosinophilic granular cytoplasm with round nucleus (f)
In addition, another tumor was noted in the sellar region with suprasellar extension, measuring 20 mm × 21 mm × 29 mm [Figure 2d and e]. Pituitary hormones were found to be within normal values. An extended right frontoparietal craniotomy was performed to approach both tumors. However, the patients’ position was suboptimal to access the sellar region limiting the resection to a partial one at this site.
Histopathology of the right frontoparietal lesion revealed a glioblastoma multiforme [Figure 2b and c] while the sellar tumor was identified as pituitary adenoma [Figure 2f]. Since his hormonal profiles were normal, nonfunctioning pituitary adenoma was designated. The postoperative period was uneventful, and the patient was sent for adjuvant therapy. Unfortunately, the patient refused adjuvant therapy and was lost to for any further follow-up.
Patient 3: Planum sphenoidale meningioma and pituitary adenoma
A 62-year-old female patient presented with progressively blurred vision in her right eye, which had occurred over the course of 1 year. On examination, she had normal visual acuity in her left eye but only 20/200 in her right eye. A cranial T1W MRI with I/V gadolinium revealed a homogeneously enhancing, board-based tumor at the planum sphenoidale with a dural tail sign, measuring 37 mm × 30 mm × 18 mm. Interestingly, a second enhancing sellar tumor with suprasellar extension was observed posterior to the former [Figure 3a and b]. Therefore, the patient underwent a subfrontal approach for microsurgical resection. The anterior tumor showed a good arachnoidal plane for dissection but was attached to the dura of the planum sphenoidale. This tumor was subtotally removed due to high vascularity.

- Case 3 – Sagittal magnetic resonance imaging showing enhanced extra-axial mass at planum sphenoidale and the mass had more extension into the suprasellar region. The pituitary gland was enlarged and heterogeneous enhancement (a). Axial magnetic resonance imaging showing the pituitary tumor is left posteriorly displaced from the planum sphenoidale tumor (b). Histological photomicrograph of the planum sphenoidale tumor showing proliferation of meningothelial cells that arrange in a whirling pattern. The tumor cells have an elongated shape with a homogeneous round nucleus (c)
Histological characteristics were those of a meningothelial meningioma. Tumor cells formed lobules where they appeared to form a syncytium with oval nuclei, occasionally showing central clearing [Figure 3c]. The tumor was immunostained and found to be positive for epithelial membrane antigen. After surgery, the patient developed Gram-negative septicemia and required antibiotic administration for 2 weeks. She recovered well and her vision, in the right eye, gradually improved too. At follow-up, she maintained independent daily activities. She does not wish to pursue a second surgery for the tumor residual.
Patient 4: Intraosseous meningioma and splenium anaplastic astrocytoma
A 61-year-old female patient presented with progressive headaches. Ten years prior, she had been diagnosed with an intraosseous meningioma at the right frontal base [Figure 4a and b] for which she underwent a right-sided craniotomy for resection. Follow-ups with MRIs were obtained annually and showed stable residual disease. However, over the course of 3 months, before admission, she had begun to develop progressively worse headaches. After a repeat of an MRI, a newly infiltrative heterogeneously enhancing mass involving bilateral splenium of the corpus callosum was observed. To obtain a pathological diagnosis, an image-guided biopsy was carried out [Figure 4c and d].

- Case 4 – Bone window axial computed tomography brain showing 1.7 cm thickening of bone involving the right sphenoid bone and superior and lateral orbital wall (a). Magnetic resonance imaging showing enhanced en plaque soft tissue and dural enhancement in the right middle cranial fossa. Severe right exophthalmos was observed (b). Magnetic resonance imaging showing 3.3 cm × 4.6 cm heterogeneously enhancing mass involving bilateral splenium of corpus callosum (c and d). Histological photomicrograph of splenium anaplastic astrocytoma showing aggregation of oval-to-elongated shape glial cell with and dysplastic nuclei (e)
Microscopic examination revealed hyperchromatic nuclei and mitotic figures. However, no areas of necrosis, hemorrhage, or endothelial hyperplasia were observed. A diagnosis of anaplastic astrocytoma WHO Grade III was made [Figure 4e]. Her hospital course was uneventful. Later, she was given a course of adjuvant external beam radiotherapy. She did not receive chemotherapy due to financial reasons. She has been followed up since, and she was still able to manage daily activities at the last follow-up.
Patient 5: Pituitary adenoma with multiple meningiomas
A 50-year-old female patient presented with a chronic headache for 3 months and with progressively worsening visual field defects. On examination, a dense bilateral temporal hemianopia with normal visual acuity was detected. Her hormonal laboratory demonstrated mildly elevated prolactin (64.5 ng/ml) although all other hormones were within normal range. Cranial MRI revealed a heterogeneously enhancing sellar tumor with suprasellar extension and chiasma compression [Figure 5a]. Two other hemispheric lesions were identified, which imposed as homogeneously enhancing solid nodules (1.1 cm on the right posterior parietal lobe and 1 cm on the left vertex with hyperostosis of adjusted skull). These were interpreted as likely meningiomas [Figure 5c and d]. The patient underwent an endoscopic endonasal transsphenoidal approach for successful sellar tumor removal.

- Case 5 – Axial magnetic resonance imaging showing a heterogeneous enhanced pituitary tumor with suprasellar extension (a). Histological photomicrograph of pituitary tumor showing proliferation of round-to-oval cell in a sheeting pattern. The tumor cells have homogeneous round nucleus fine granular chromatin (b). Two extra-axial enhanced nodules at the left vertex and posterior parietal lobe (c and d)
Microscopic examination of this surgical specimen showed epithelioid cells with a sheet-like growth pattern. A histological diagnosis of pituitary adenoma was made [Figure 5b]. The postoperative period was without incident. The patient is being followed clinically and expectantly for her convexity lesions. Her visual field defects have improved since.
Patient 6: Pineal epidermoid cyst with vestibular schwannoma
A 37-year-old female was referred from a provincial hospital due to a rapidly, worsening headache over a period of 2 weeks. According to her history, she had been suffering from progressive headaches for 5 years as well as from hearing disturbances on the right side for 1 year. Two weeks before admission, her headaches got rapidly more severe and she developed a bilateral papilledema. Cranial MRI revealed two tumors located in the pineal region and at the cerebellopontine angle (CPA), causing obstructive hydrocephalus.
A cranial T1W MRI revealed a nonenhancing large tumor at the pineal region with third ventricle extension. This tumor displayed restricted diffusion on diffusion-weighted MRI [Figure 6a and b]. The second lesion imposed as a homogeneously enhancing cone-shaped tumor at CPA at the level of the right internal acoustic canal. The findings were compatible with vestibular schwannoma [Figure 6d].

- Case 6 – Sagittal magnetic resonance imaging revealing a lobulated extra-axial cystic lesion about 6.3 cm × 6.6 cm × 4.2 cm located at suprapineal recess and superior cerebellar recess. The lesion insinuates into posterior aspect of the third ventricle (a). Diffusion-weighted imaging showing restricted diffusion pineal lesion (b). The tumor shows proliferation of benign squamous epithelium with keratin material (c). Extra-axial heterogeneous enhancing mass is located at the right cerebellopontine angle with right internal acoustic canal invasion. Widened right internal auditory canal is observed (d)
The patient first underwent ventriculoperitoneal shunting for diversion of her cerebrospinal fluid. Later, an occipital transtentorial approach was chosen for pineal tumor removal. Near total tumor removal was achieved and the postoperative period was uneventful.
Histologically, the microscopic examination showed epithelium and lamellated keratin [Figure 6c], and the final diagnosis was epidermoid tumor. At last follow-up, she had recovered well and decided to postpone any CPA tumor surgery.
DISCUSSION
In this case series, the authors report these six cases of coexisting, primary brain tumors observed in individual patients. They then reviewed the available literature for similar reported cases of coexistence between various tumor entities.
Clinical features
The clinical characteristics of the patient cohort are summarized in Table 1. In addition, the summary table of all cases that have been reportedly been performed is in Appendix 1. This rare scenario is more frequently observed in the fourth-to-sixth decades of life, and we found a female predominance. The most frequently encountered combination is meningioma and pituitary adenoma.

The majority of coexisting tumors occurred simultaneously and in distinct locations. Almost a half of the tumors found under these circumstances were meningiomas. In cases of pituitary lesions, they were nonfunctioning adenomas.
Other associations with meningioma were described as glioma, primary cerebral lymphoma, schwannoma, and craniopharyngioma. In cases in which gliomas were found in this condition these where usually high grade glioma. Interestingly, an unusual combination between an epidermoid cyst and a schwannoma was found in our own series.
From pooled analysis, the mortality rate has been reported to be 21.1% (15/71). Mean survival is 10.36 months; however, the reason of death was restricted from secondary citation. Notice, 86.6% (13/15) of lethal cases are a combination between glioma and meningioma and 66.7% (10/15) are malignant glioma.
Pathogenesis
Various hypothesis of an association between the “tumor pairs” has been advocated in the past. Local tissue irritation from perilesional edema caused by the first tumor has been considered as a factor in inducing astrocyte or arachnoid cell transformation, causing neoplastic proliferation in the future.[1633] However, this hypothesis cannot completely explain the observations made since more than half of the simultaneously existing tumors are topographically disconnected (spatially remote) lesions. Therefore, another hypotheses have been proposed where a common genetic pathway is suggested, including the p53 disruption and receptor tyrosine kinase signaling molecules that have an expression of platelet-derived growth factor receptors (PDGFRs).[34]
PDGF is one of the numerous growth factors that appears to play a significant role in angiogenesis and that can lead to tumorigenesis.[4041] Suzuki et al. reported an association of glioblastoma simultaneously presenting with meningioma. In their study, both tumors were strongly positive for PDGFR expression levels as seen through immunohistochemistry.[34] PDGFR subunits were classified in two types: alpha- and beta-subunits.
The alpha-subunit of the human PDGFR-alpha (PDGFRA) gene maps on q11-q12 of the human chromosome 4[42] and gene encoding the beta-subunit of the human PDGFR-beta (PDGFRB) maps on human chromosome 5 at region 5q31-32.[43] Theoretically, coexisting tumors should therefore carry abnormalities of chromosome 4 or 5, which PDGFRA and PDGFRB are transcribed.[40414243]
Nevertheless, relevant studies did not demonstrate concordant evidence. Nestler et al. undertook chromosomal and genetic investigations in an assumed association between glioblastoma multiforme and meningioma. However, chromosomal analysis from peripheral blood cells of their patient was normal, with an absence of chromosome 17p deletions (p53). Beyond that, genetic analysis of tumor cells, using comparative genomic hybridization (CGH), found monosomy of chromosome 22 in meningioma cells and amplification of chromosome 7 and loss on chromosome 10 in glioblastoma cells.[30] Similarly, Ohba et al. recently reported the results of CGH of tumor cells in which losses on 22q and gain 3, 5, 7, 11, 12, 13q, 15q, 17q, 20, and 21q in meningioma cell versus losses on 1p35, 9pter-21, 10, 11q23, 13q, 14q, 20q, and 22q and gain on 7 in glioblastoma cells.[31] Since molecular variations isolated each tumor and were not linked with chromosome 4 or 5 alterations, there has been inconclusive common pathways, which carried out etiology of coexisting tumors.
We, therefore, believe that the concept of an association of multiple, primary brain tumors is incompletely supported and the current evidence does not allow us to conclude the existence of a common genetic pathway to explain pathogenesis of this condition. However, such investigations are severely limited because of this being such a scarce scenario, with only a few patients reported in the literature as of now.
In epidemiology, those of the female gender coupled with increasing age are at greater risk for meningioma while glioma is frequently observed in the mid-30s of male gender. Vernooij et al. conducted MRIs of the brain in 2000 people to determine the prevalence of incidental brain findings. The prevalence of incidental brain tumor was 1.6%. Meningioma was the most common tumor (0.9%) while pituitary adenoma could incidentally be found in 0.3%.[6] The “Swiss cheese model” of accident causation explained that various errors fortuitously become accidents and incidents.[4445] Adaptively, the concept of truly incidental events should be considered like a model.
Management issues
According to the literature, surgery is usually the treatment of choice for this scenario, but the decision to operate on both tumors at the same time is debatable. Simultaneous and adjacent tumors are found in only 42.2% (30/71) of cases. A single operation could be accepted in cases when both tumors are spatially contiguous.[21] However, 49.2% (29/59) of stimulus cases show discontinuous tumors. Hence, the question will come up: “what tumor should be removed first?” We propose the idea of management for this scenario focus on the natural history of the disease.
Decision-making based on the presumed histopathology is an option. If a glioma is assumed, it will be the key determinant of the patients’ prognosis in this scenario because 73% (19/26) of gliomas are malignant while almost all of the other coexisting tumors are benign. Planning of any therapy would, therefore, focus on the glioma. This correlates well with the observation that 86.6% (13/15) of reported mortality occurred in patients in whom one of the lesions was a glioma. It is, therefore, our opinion that the malignant tumor should be removed first and treatment should center around the best, adjuvant management.
Hence, a second operation for the removal of any remaining lesions can be delayed or such lesions can also be observed only when these secondary tumors are likely benign, asymptomatic, small sized, or slow to grow.
CONCLUSION
The authors described a case series of rare patients, who presented with coexisting, primary brain tumors. The authors presented their patients in detail and summarized other clinical features found in the literature.
The pathomechanism of this rare disease scenario has been inconclusive. Further genetic investigations performed on tumors found in this scenario will help to answer the question whether there is a common underlying genetic pathway for both (coexisting) tumors. As of now, surgical management remains a mainstay of treatment. In cases in which high-grade glioma had been found as one of the lesions, it is evident that it conveys a poor prognosis.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
The authors would like to offer their special thanks to Dr. Pornprot Limprasert and Kanut Thapasathitsin for their advice on information in regard to human genetics.
REFERENCES
- Radio-induced gliomas: 20-year experience and critical review of the pathology. J Neurooncol. 2008;89:169-77.
- [Google Scholar]
- Meningiomas. Their Classification, Regional, Behavior, Life History and Surgical End Results. Baltimore: Charles C Thomas; 1938. p. :506-7.
- [Google Scholar]
- CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the united states in 2007-2011. Neuro Oncol. 2014;16(Suppl 4):iv1-63.
- [Google Scholar]
- Incidental intracranial findings and their clinical impact; The HUNT MRI study in a general population of 1006 participants between 50-66 years. PLoS One. 2016;11:e0151080.
- [Google Scholar]
- Incidental findings on brain MRI in the general population. N Engl J Med. 2007;357:1821-8.
- [Google Scholar]
- Sarcoma arising in irradiated bone: Report of eleven cases 1948. Cancer. 1998;82:8-34.
- [Google Scholar]
- The association of meningioma and pituitary adenoma: Report of seven cases and review of the literature. Eur Neurol. 1993;33:416-22.
- [Google Scholar]
- Simultaneous meningioma and glioma. Difficulties of neuroradiological diagnosis. Report of a case. Ital J Neurol Sci. 1983;4:481-3.
- [Google Scholar]
- Coexistence of growth hormone-secreting pituitary adenoma and intracranial meningioma: A case report and review of the literature. J Endocrinol Invest. 1993;16:703-8.
- [Google Scholar]
- A case of coexisting cerebellopontine angle meningioma and schwannoma. Neurol India. 2000;48:198.
- [Google Scholar]
- MRI finding of simultaneous coexistence of growth hormone-secreting pituitary adenoma with intracranial meningioma and carotid artery aneurysms: Report of a case. Pituitary. 2007;10:299-305.
- [Google Scholar]
- Pituitary adenoma associated with intraventricular meningioma: Case report. Skull Base. 2007;17:347-51.
- [Google Scholar]
- Intracranial meningioma and astrocytoma in the same patient. Case report and review of the literature. J Neurosurg Sci. 1995;39:27-35.
- [Google Scholar]
- Concurrent adjacent meningioma and astrocytoma: A report of three cases and review of the literature. Neurosurgery. 1995;36:599-604.
- [Google Scholar]
- Contiguous synchronous occurrence of primary cerebral lymphoma and meningioma. Br J Neurosurg. 2007;21:35-8.
- [Google Scholar]
- Meningioma interdigitated with primary central nervous system B-cell lymphoma: A case report and literature review. Surg Neurol Int. 2011;2:181.
- [Google Scholar]
- Simultaneous occurrence of meningioma and glioma in brain: Report of two cases. J Clin Neurosci. 2003;10:252-4.
- [Google Scholar]
- The coexistence of pituitary adenomas and meningiomas: Three case reports and a review of the literature. Br J Neurosurg. 1989;3:59-69.
- [Google Scholar]
- Combined occurrence of primary cerebral lymphoma and meningioma. Neurosurg Rev. 1995;18:45-8.
- [Google Scholar]
- Three distinct co-existent primary brain tumors in a patient. J Cancer Res Ther. 2009;5:293-6.
- [Google Scholar]
- Coexistence of intracranial meningioma and primary malignant lymphoma – case report. Neurol Med Chir (Tokyo). 1990;30:268-71.
- [Google Scholar]
- Two primary brain tumors, meningioma and glioblastoma multiforme, in opposite hemispheres of the same patient. J Clin Neurosci. 2002;9:589-91.
- [Google Scholar]
- Endoscopic endonasal transsphenoidal approach for resection of a coexistent pituitary macroadenoma and a tuberculum sellae meningioma. Asian J Neurosurg. 2014;9:236.
- [Google Scholar]
- Simultaneous presentation of meningiomas with other intracranial tumours. Br J Neurosurg. 2005;19:368-75.
- [Google Scholar]
- Pituitary adenoma and parasagittal meningioma: An unusual association. Neurol India. 2000;48:72-4.
- [Google Scholar]
- Glioblastoma simultaneously present with meningioma – Report of three cases. Zentralbl Neurochir. 2007;68:145-50.
- [Google Scholar]
- A glioblastoma arising from the attached region where a meningioma had been totally removed. Neuropathology. 2011;31:606-11.
- [Google Scholar]
- Two distinct intracranial tumors of different cell types in a single patient. Case report and review of the literature. Neurosurg Rev. 1994;17:305-8.
- [Google Scholar]
- Intracranial meningiomas associated with glial tumours: A review based on 54 selected literature cases from the literature and 3 additional personal cases. Acta Neurochir (Wien). 1991;110:133-9.
- [Google Scholar]
- Glioblastoma simultaneously present with adjacent meningioma: Case report and review of the literature. J Neurooncol. 2010;99:147-53.
- [Google Scholar]
- Two distinct primary brain tumors, in same region of the same patient: A case report. J Neurooncol. 2006;79:219-20.
- [Google Scholar]
- Coincidental pituitary adenoma and parasellar meningioma: Case report. Neurosurgery. 1986;19:267-70.
- [Google Scholar]
- Value of petrosal sinus sampling: Coexisting acromegaly, empty sella and meningioma. Neuroradiology. 2004;46:1027-30.
- [Google Scholar]
- Pituitary adenoma and meningioma in the same patient. Report of three cases. Eur Arch Psychiatry Neurol Sci. 1989;238:144-8.
- [Google Scholar]
- Platelet-derived growth factor expression and stimulation in human meningiomas. J Neurosurg. 1994;81:388-93.
- [Google Scholar]
- Platelet-derived growth factor and its receptors in human glioma tissue: Expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992;52:3213-9.
- [Google Scholar]
- The human PDGF receptor alpha-subunit gene maps to chromosome 4 in close proximity to c-kit. Hum Genet. 1990;85:383-5.
- [Google Scholar]
- Handbook of Growth Factors. Hematopoeitic Growth Factors and Cytokines. Vol 3. Florida: CRC press; 1994. p. :297-78.
- [Google Scholar]
- The swiss cheese model of safety incidents: Are there holes in the metaphor? BMC Health Serv Res. 2005;5:71.
- [Google Scholar]
Appendix 1
Summary of 71 cases of coexisting glioma and meningioma without previous radiotherapy or phakomatosis in English literatures





