
Translate this page into:
Morphological spectrum of peripheral nerve sheath tumors: An insight into World Health Organization 2013 classification
This is an open access article distributed under the terms of the Creative Commons Attribution NonCommercial ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Introduction:
Peripheral nerve sheath tumors (PNSTs) are neuroectodermal in origin. Now these tumors are classified under World Health Organization (WHO) classification of tumors of soft tissue and bone 2013.
Objective:
To study the morphological spectrum of PNST and to study the secondary degenerative changes associated with it.
Materials and Methods:
This study was conducted from January 2010 to June 2015. The gross details of tumor and patient's demographic profiles were reviewed. The hematoxylin and eosin stained slides were reassessed and the lesions were categorized and classified as per the WHO 2013 classification. The tumors were also assessed for secondary degenerative changes.
Results:
Our study comprised 143 cases of PNST. Age of the patients ranged from 5 to 75 years. 21–30 years is the most common age of occurrence with head and neck being the most common site. The PNSTs observed in the present study were neurofibroma (NF) (61.5%), schwannoma (36%), malignant PNST (2%), and granular cell tumor (0.5%). Nearly 10% of NF fulfilled the criteria for neurofibromatosis 1 (NF1). Rare tumors such as plexiform schwannoma and granular cell tumor were also observed. Malignant tumors were larger in dimension than benign. Myxoid, cystic, and hyaline changes were commonly associated with benign tumors while necrosis, hemorrhage, and mitotic activity were seen with malignant tumors.
Conclusion:
This series highlights the pathological variants of PNST along with their morphological changes and NF1 association. It is essential to be familiar with all these variants of PNST for accurate diagnosis as they have varied biological behavior.
Keywords
Granular cell tumor
malignant peripheral nerve sheath tumor
neurofibroma
neurofibromatosis
peripheral nerve
schwannoma

Introduction
Peripheral nerve sheath tumors (PNST) are neuroectodermal in origin.[1] They consist of spectrum of tumors ranging from benign tumors such as neurofibroma (NF) and schwannoma to malignant tumors, called malignant PNST (MPNST).[2] More than 90–95% of PNST are benign. MPNST is a rare tumor with incidence of 0.001% and in patient of neurofibromatosis 1 (NF1), its incidence increases to 4–4.6%.[1] Initially, World Health Organization (WHO) has classified these tumors under 2007 WHO classification of tumors of the central nervous system under cranial and peripheral nerves, skin, head, and neck.[345] Now these tumors are classified under 2013 WHO classification of tumors of soft tissue and bone thus bringing all mesenchymal tumors under a single roof.[6] Literature review revealed only few articles describing the spectrum of PNST.[12] Benign PNSTs (BPNST) are not studied much while morphological, immunohistochemical, and molecular studies on MPNST are available.[7891011] In this communication, we describe morphological spectrum of 143 PNST from a single center of South India.
Materials and Methods
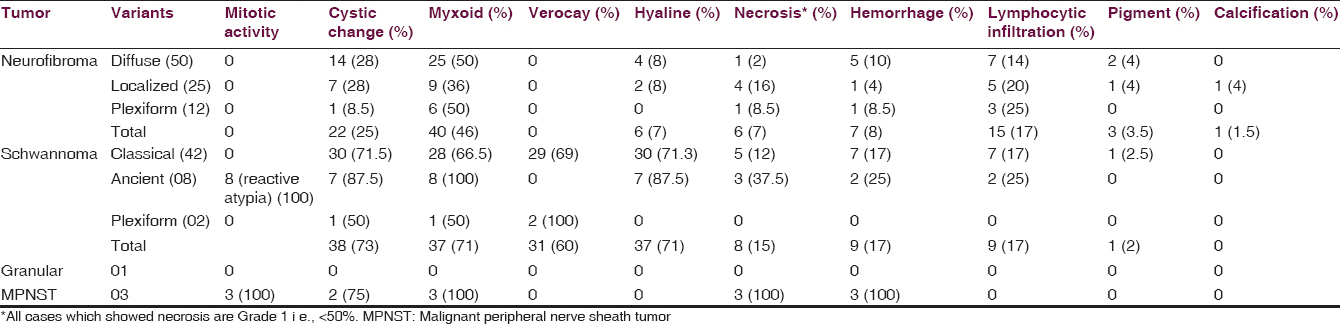
This is a retrospective study conducted at a Medical College and Teaching Hospital for 6½ years from January 2010 to June 2015. Histopathologically diagnosed cases of PNSTs during the study period were included. The demographic profiles of the patients such as age, sex, and site of tumor and gross findings such as size and other details were noted from the pathology case files. The hematoxylin and eosin (H and E) stained slides were retrieved from the archives and in cases where H and E slides are not available, paraffin block was removed and 3–5 µ thick sections were cut and stained by H and E. The slides were reassessed and the lesion were categorized and classified as per the WHO 2013 classifications.[6] The tumors were assessed for morphological features such as (a) variants, (b) mitotic activity, (c) necrosis, (d) cystic change, (e) verocay body, (f) hemorrhage, (h) myxoid change, (i) pigment, and (j) calcification.
The diagnosis of MPNST was made on the criteria proposed by Weiss and Goldblum.[12] Necrosis was graded as 0, 1, and 2 representing 0%, <50%, and >50%, respectively. Malignant tumors were graded as low grade and high grade, based on mitotic activity, tumors with mitotic count more than 5/HPF are considered as high grade.[11] For the diagnosis of NF1, the criteria given in 2007 WHO classification of tumors of the central nervous system were used.[13]
Results
The present study comprised 143 cases of PNSTs ranging in age from 5 to 75 years. These tumors were most commonly encountered in the age group of 21–30 (52 [36.5%]) years followed by 31–40 years (35 [24.5%]). Head and neck (47 [33%]) was the most common location followed by upper extremities (35 [24.5%]) and lower extremities (32 [22.5%]). Acoustic neuroma/vestibular schwannoma was not observed in the present study as this location requires neurosurgical approach for resection, same is not available in our center. The PNSTs observed in the present study in the decreasing order of frequency are NF (87 [61.5%]), schwannoma (52 [36%]), MPNST (3 [2%]), and granular cell tumor (1 [0.5%]) [Table 1]. Malignant tumors were larger in size compared to benign tumors. Schwannomas were well-encapsulated tumors while NF was poorly capsulated. Gross features of PNSTs are shown in Table 2.

Among NF, diffuse variant was most common (50 [57.5%]) followed by localized (25 [29%]) and plexiform (12 [14%]). Diffuse and localized variant are most commonly observed in second decade and head neck and extremities are most commonly involved sites. Plexiform NF was more common in multiple locations and most common age of occurrence was in the first and second decade [Table 1]. Diffuse NF had infiltrative type of growth and invades the surrounding structures while localized NF had localized type of growth. Myxoid change was the most common secondary degeneration associated with NF followed by cystic change and lymphocytic infiltration [Figure 1a–i and Table 2]. Of 90 cases of NF, 9 (10%) fulfilled the criteria for NF1, of 9 cases 8 were plexiform NF and 1 was multiple NF; the common associated finding was café au spots, in addition, one case of plexiform NF had family history.
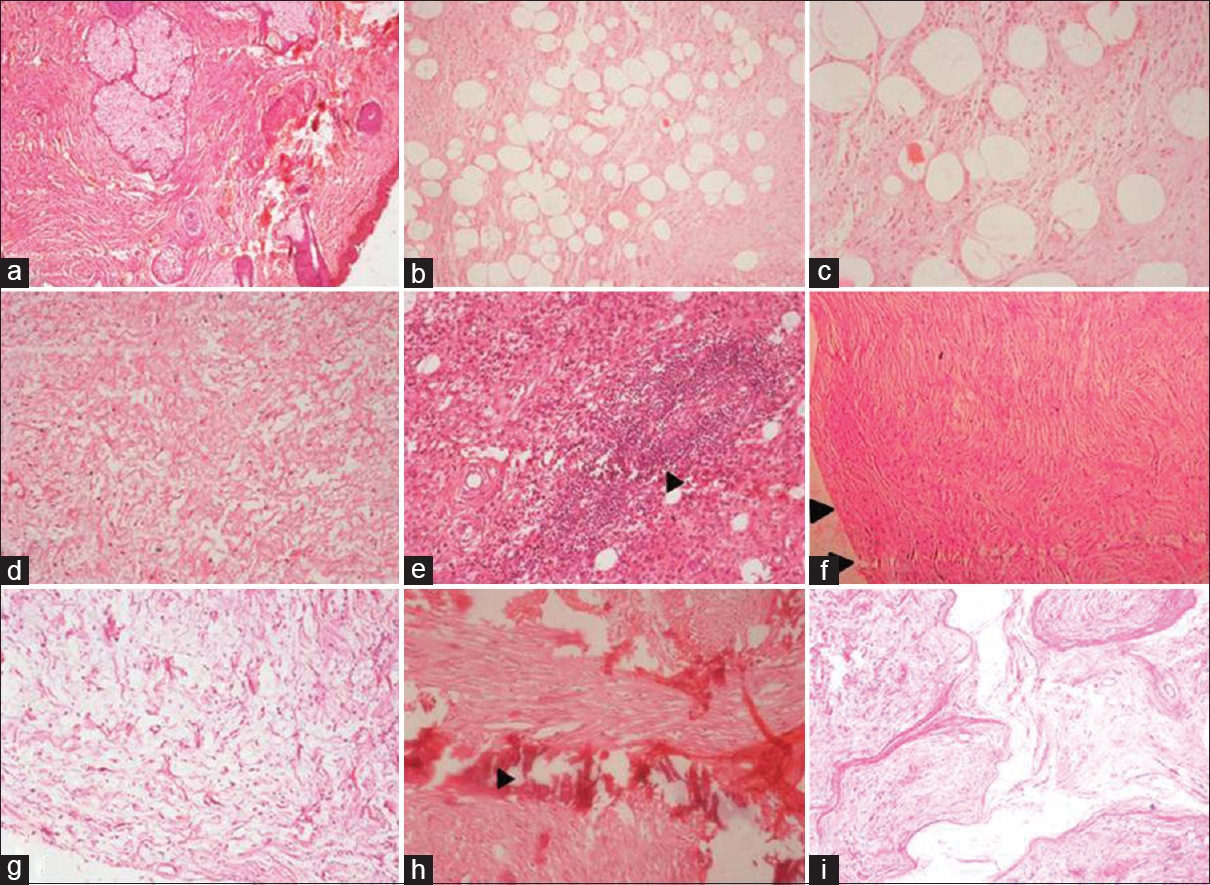
- (a-c) Microphotograph of diffuse neurofibroma, note the infiltration of tumor to dermis and subcutaneous tissue. (H and E, ×40), (d) diffuse neurofibroma showing myxoid change (H and E, ×40), (e) diffuse neurofibroma with prominent lymphoid aggregates (arrowhead) (H and E, ×40), (f) microphotograph of localized neurofibroma, note the well delineated tumor (H and E, ×40), (g) localised neurofibroma with extensive myxoid change (H and E, ×40), (h) LNF with areas of calcification (H and E, ×40), (i) plexiform neurofibroma showing tortuous and expanded nerve bundles (H and E, ×40)
Like NF, schwannomas were also most commonly located in head and neck extremities and in contrast, they are observed a decade later. Classic variant is most common (42 [81%]) followed by ancient (8 [15.5%]) and plexiform (2 [4%]) [Table 1]. Classical schwannomas have both hypercellular (Antoni A) and hypocellular (Antoni B) areas in varying proportions. Ancient variant showed predominantly hypocellular areas. The characteristic verocay bodies are seen in 100% (2 cases) of plexiform variant and 69% (25 cases) of classical schwannoma. None of ancient schwannoma showed verocay bodies. Compared to NF, secondary degenerative changes were more evident in schwannomas more so in ancient variant [Table 3 and Figure 2a–i].
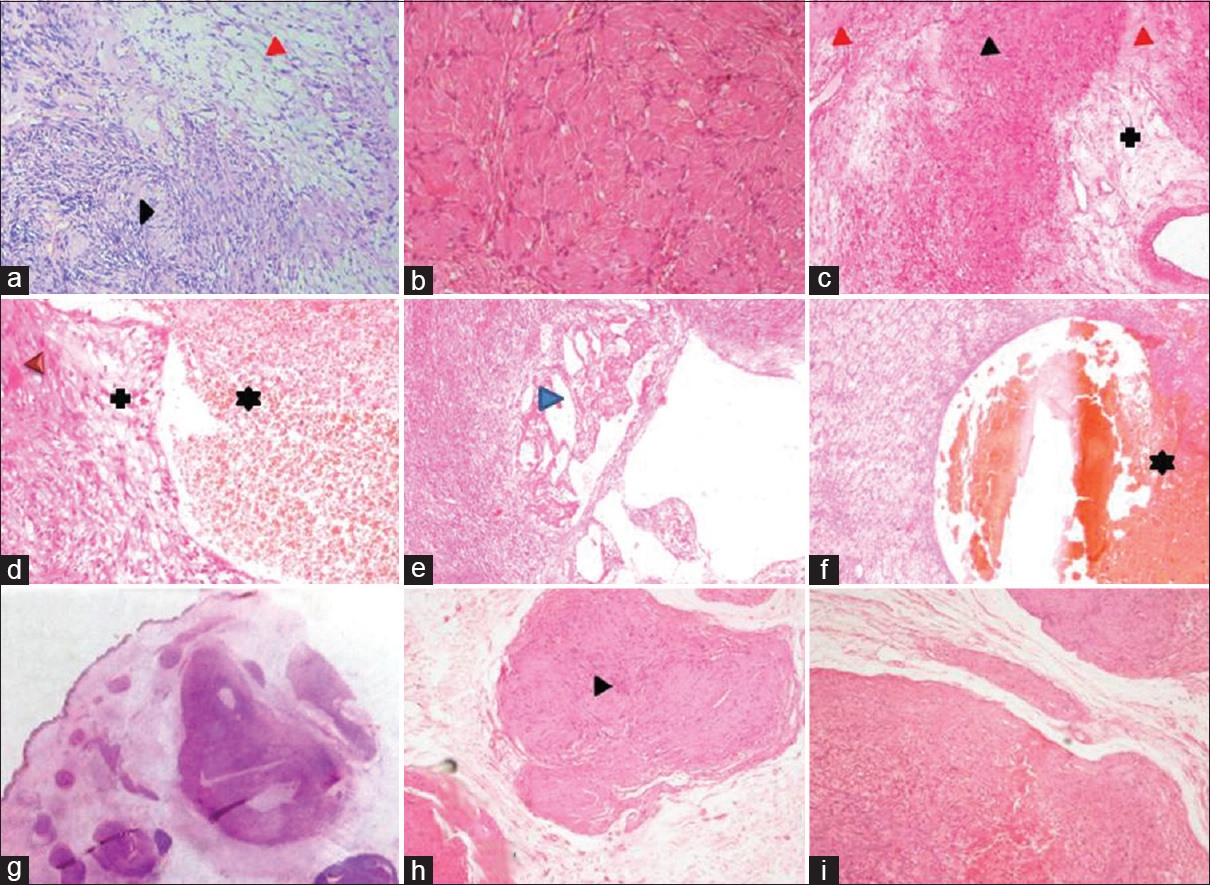
- (a) Microphotograph of conventional schwannoma showing hypercellular, Antoni A areas (black arrowhead) and hypocellular Antoni B areas (red arrowhead). (b) Schwannoma with extensive hyalinized areas. (c-f) Microphotograph of “ancient” schwannoma showing degenerative nuclear atypia (black arrowhead), hyalinized areas (red arrowhead), cystic change (blue arrow head), areas of hemorrhage (*) and myxoid change (+) (H and E, ×40). (g) whole mount section of plexiform schwannoma note the subcutaneous multiple nodules (H and E, ×4), (h and i) higher magnification of the same showing hypocellular and hypercellular areas with verocay body (black arrow)
Only one case of granular cell tumor has been observed in the present study in a 35-year-old lady, and the tumor was located in left breast. On periodic acid–Schiff (PAS) stain, PAS-positive granules were noted. On immunohistochemistry (IHC), tumor cells were positive for S-100 and vimentin [Figure 3a–f].

- (a) Microphotograph of granular cell tumor showing tumor cells arranged in interlacing pattern with benign nuclei and granular cytoplasm (H and E, ×10). (b and c) Higher magnification of granular cell tumor (H and E, ×40). (d) periodic acid–Schiff stain showing the periodic acid–Schiff positivity of cytoplasm of tumor cells (black arrow) (periodic acid–Schiff, ×40). (e) Tumor cells showing intense cytoplasmic and nuclear positivity for S-100 (immunohistochemistry, S-100, ×40) (f) tumor cells showing cytoplasmic positivity for vimentin (immunohistochemistry vimentin, ×40)
Three cases (2%) of MPNST were observed in the present study. In contrast to BPNST, MPNST was seen in fourth to fifth decade. All the cases showed mitotic activity in the range of 0–5/HPF (low grade), myxoid change, hemorrhage, and Grade 1 necrosis [Table 1]. On IHC, two cases were positive for vimentin, S-100 and negative for SMA while one case was S-100 negative, vimentin and epithelial membrane antigen positive [Figure 4a–d]. No high-grade tumors were observed in the study.

- (a) Microphotograph of malignant peripheral nerve sheath tumors showing malignant tumor cells with hyperchromatic, pleomorphic nuclei with eosinophilic to clear cytoplasm (H and E, ×40) (b) tumor cells showing diffuse cytoplasmic and nuclear positivity for S-100 (immunohistochemistry S-100, ×40). (c) Tumor cells showing cytoplasmic positivity for vimentin (immunohistochemistry vimentin, ×40). (d) Tumor cells showing nuclear positivity for ki 67 indicating proliferative activity (black arrowhead) (immunohistochemistry ki 67, ×40)
Discussion
With the invention of IHC and molecular diagnostic methods, many tumors are now getting proven to be neural in origin, hence, the list of PNST are increasing. To the existing list of WHO classification, new variants, many benign, and malignant tumors were added and classified under soft tissue tumors. Benign tumors which are added are Granular cell tumor, dermal nerve sheath myxoma (DNM), solitary circumscribed neuroma (SCN), ectopic meningioma/meningothelial hamartoma, nasal glial heterotopias, benign triton tumor (BTT), and hybrid nerve sheath tumors. Malignant triton tumor, malignant granular cell tumor (MGCT), and ectomesenchymoma (EM) are the malignant tumors now proven to be of neural origin.[6]
NF and schwannoma are the well-defined benign tumors of PNST. Schwann cells are considered by many authors as the cell of origin for both the tumors; however, dermal NF may arise from neural crest cell.[12] An ultrastructural study by Erlandson and Woodruff[14] on 23 schwannomas, 10 NFs, and 10 MPNST concluded that schwannoma to be consisting of well-differentiated Schwann cells; hence, the cell of origin is a Schwann cell. The neoplastic cell in NF did not resemble Schwann cell; on contrary, in six cases, it resembled perineural cells. NF also consists of nonneoplastic cells such as fibroblast and axon. Hence, NF and schwannomas are distant entities. Controversy exists in the literature regarding the origin of MPNST. Few authors believe that MPNST is the malignancy arising from schwannoma and designate them as malignant schwannoma. Few authors believe that they arise from neural crest cells with neural differentiation.[2]
NF occurs commonly in the age group of 20–30 years with head and neck being common site (as observed in our study). Among the variants of NF, localized is the most common followed by diffuse and plexiform; however, in our study, we have observed diffuse variant as most common followed by localized and plexiform.[12] Diffuse variant presents as plaque-like enlargement of nerve while localized present as fusiform enlargement of nerve [Figure 5a]. Plexiform arise from major nerve or trunk and appearance called “bag of worms,” [Figure 5b] microscopically it consists of admixture of diffuse and localized areas.[215] NF is well delineated and poorly encapsulated. Secondary degenerative changes are uncommon with NF.[12] In a study by Gabhane et al.,[1] capsule was evident in 14% of cases, myxoid change was observed in 18%, and pigment in 5.5%. In the present study, 8% of NF was capsulated and secondary degenerative changes such as myxoid change (46%), cystic change (25%), and lymphocytic infiltration (17%) were more evident in all variants.
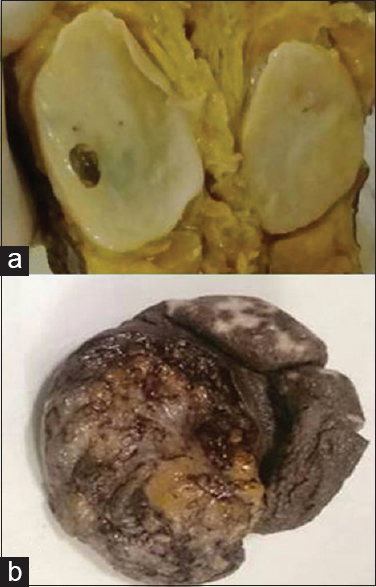
- (a) Gross photograph of localized neurofibroma showing fusiform growth with myxoid and cystic areas. (b) Gross photograph of plexiform neurofibroma with thickened skin and gray-white tumor (Bag of worms)
Like NF, schwannoma is also common in head and neck region but they are observed a decade later, i.e., 20–50 years (as observed in our study).[12] In contrast to NF, schwannomas are capsulated, secondary changes are more evident, and variants are well defined and practiced by a pathologist. Conventional or classic schwannomas are most common followed by cellular, ancient, plexiform, and melanotic.[6] To the existing list, WHO 2013 added microcystic/reticular schwannoma.[16] Classical schwannomas are easy to diagnose, and they have alternating areas of hypercellular areas, consisting of palisading arrangement of nuclei of spindle cells forming verocay body and hypocellular areas consisting of myxoid material, macrophages, and cystic change. Hyalinized blood vessels are constant findings.[216]
Cellular variant will have more hypercellular areas with minimal hypocellular areas but verocay bodies are absent. In ancient variant, secondary degenerative changes are more evident along with extreme degree of hyalinization. Verocay bodies are usually absent.[21617]
Plexiform variant is rare and less commonly associated with NF2. The tumor usually occurs in subcutaneous tissue; however, deep visceral sites are also reported. These tumors arise from nerve plexuses or fascicles, less circumscribed and unencapsulated (as observed in our cases). Microscopically, they contain predominantly Antoni A areas. They have low malignant potential and local recurrence is high.[1618]
Melanotic schwannoma (MS) is a rare variant of schwannoma, and the tumor arises from cervical and thoracic spinal root. Microscopically, they consist of areas of spindle and epithelioid cells with melanin pigment. Sometime, in addition, they show calcification termed, “Psammomatous MS,” which are associated with carney complex. MS has high malignant potential but rarely metastasize.[192021] Microcystic/reticular schwannoma occurs in the visceral organs with gastrointestinal tract being the most common; however, these tumors are reported in other sites. Microscopically, they have microcyst-rich network of interconnected spindle cells.[16222324] In the present study, we have observed conventional, ancient, and plexiform variants.
Secondary degenerative changes are common in schwannoma. In a study by Gabhane et al.,[1] observed secondary degenerative changes such as cystic change, hyalinization, necrosis, myxoid change, and verocay bodies were observed in 85.18%, 72.22%, 59.25%, 24.07%, and 70.37% cases, respectively. In the present study, we have observed cystic change, hyalinization, necrosis, myxoid change, and verocay bodies in 73%, 71%, 15%, 71%, and 60% cases, respectively, and our findings are in consistent with literature.
Unlike BPNST, MPNST is rare (0.001%) and the age group observed ranged from 16 to 60 years with extremities being the most common site (as observed in our study).[12252627] Like BPNST, even MPNST is associated with secondary degenerative changes. In a study, Gabane et al.[1] observed necrosis and hemorrhage in 75% of cases and cystic change in 50% of tumor. In the present study, myxoid change, necrosis, and hemorrhage were noted in 100% of cases and cystic change in 75% of cases.
The common differential diagnoses of MPNST include other mesenchymal tumors such as rhabdomyosarcoma, leiomyosarcoma, synovial sarcoma, fibrosarcoma, and malignant fibrous histiocytoma.[12] Hence, it is justifiable to include MPNST under soft tissue tumors. Mean height × width of MPNST was more compared to BPNST. The comparison of dimensions of BPNST and MPNST with the previous literature is shown in Table 4.[126]

Diagnosis of schwannoma and NF is straight forward and requires IHC rarely. S-100 is found to be constantly expressed in schwannoma and NF.[1226] As the differential diagnoses of MPNST are wide, IHC is essential for confirmation. S-100 which is found to be constantly expressed in BPNST may not be positive in all cases of MPNST[926] (as observed in one of our case). Nestin is an intermediate filament of neuroectodermal stem cells. Nestin is constantly expressed in MPNST and is a sensitive marker. It is also expressed in rhabdomyosarcoma, malignant melanoma, and leiomyosarcoma. It is negative or weakly expressed in leiomyoma, NF, schwannoma, synovial sarcoma, liposarcoma, carcinosarcoma, and malignant fibrous histiocytoma.[9] In difficult cases of BPNST and MPNST, proliferating cell nuclear antigen and Ki67 help in diagnosis.[28]
Perineuroma is a benign tumor showing perineural differentiation. These tumors are most commonly seen in middle age with extremities being the most common site and are rarely associated with NF1.[29] Granular cell tumor is now proved to be neuroectodermal in origin. These tumors are most common in head and neck region and are component of Noonan syndrome. Microscopically, these tumors show granular eosinophilic cytoplasm, hence the name.[303132] We have observed only one case of granular cell tumor in 35-year-old female in left breast.
DNM most commonly occurs in skin and subcutaneous tissue of extremities. DNM is characterized by multinodular growth pattern, bordered by collagen. The neoplastic spindle cells are embedded in a myxoid matrix. It is known for recurrence if incompletely resected.[33] SCN is most commonly observed in adult population and head and neck being the usual site of occurrence. In contrast to NF and schwannoma, in SCN, Schwann cells replete with axon which is neurofilament-positive.[34]
Ectopic meningioma is a meningioma occurring outside the normal anatomical region. Except for the site these tumors behaves like other meningioma. Ectopic meningioma needs to be differentiated from hamartomatous condition which contains nonneoplastic proliferation of arachnoidal cells.[35] Nasal glial heterotopias are the presence of mature glial tissue outside the cranial cavity, most commonly observed in nasal cavity of children.[36] BTT is extremely rare, and they contain mature skeletal muscle with clusters of nerve fibers.[37]
The interesting tumor added to the newer classification is hybrid BPNST. They consist of discrete areas of more than one histological type. The common combinations are NF-schwannoma, schwannoma-perineuroma, and NF-perineuroma. NF-schwannoma is the most common and associated with schwannomatosis and neurofibromatosis. IHC is required for diagnosis of these mixed tumors.[38]
MGCT and EM are newly added malignant tumors. MGCT is a high-grade sarcoma most commonly occurs in extremities and head and neck region. These tumors arise from Schwann cell and have abundant granular cytoplasm.[39] EM is a variant of rhabdomyosarcoma with neural component.[40]
NF1 and NF2, schwannomatosis, and Noonan syndrome are genetic syndromes associated with PNST. NF1 is a neurocutaneous disease affecting 1 in 3000 people and transmitted as autosomal dominant disorder. The defective gene is located on chromosome 11.[1] All variants of NF can be seen in patients with NF1 but plexiform variant is most common. NF in NF1 is multiple and segmental.[12] Gabhane et al.[1] observed that 7.93% of BPNST and 1.58% of MPNST are associated with NF1. In the present study, 10% of NF is associated with NF1 and plexiform variant is the most common. NF2 also transmitted as autosomal dominant disorder occurs due to the germ line mutation of NF2 gene on chromosome no 22. Sporadic mutation of NF2 is termed as schwannomatosis. Both syndromes are rare with an incidence of 1:40,000–1:80,000, respectively. Both conditions will have peripheral nerve and cranial nerve schwannomas; in addition, NF2 will have other intracranial tumors.[41] Noonan syndrome is a multisystem disorder transmitted as autosomal dominant pattern. The disorder is due to the alteration in RAS–MAPK pathway and granular cell tumors are a component of it.[42]
Conclusion
Due to the modern diagnostic modalities, list of PNST is increasing. In the present study, we have described the gross and microscopic features of benign and MPNSTs and their variants. As these tumors have varied biological behavior and few are associated with inherited tumors, pathologists must be aware of all these variants for accurate diagnosis.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Morphological spectrum of peripheral nerve sheath tumors: A series of 126 cases. Indian J Pathol Microbiol. 2009;52:29-33.
- [Google Scholar]
- Pathology of peripheral nerve sheath tumors: Diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012;123:295-319.
- [Google Scholar]
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, eds. WHO Classification of Tumours of the Central Nervous System. Lyon: IARC; 2007. p. :8-9.
- Barnes L, Eveson JW, Reichart P, Sidransky D, eds. World Health Organisation Classification of Tumours of the Head and Neck Tumours. Lyon: IARC; 2005. p. :10.
- World Health Organization Classification of Tumours. In: LeBoit PE, Burg G, Weedon D, Sarasain A, eds. Pathology and Genetics of Skin Tumours. Lyon: IARC Press; 2006. p. :9-10.
- [Google Scholar]
- Fletcher CD, Bridge JA, Hongendroorn PC, Mertens F, eds. WHO Classification of Tumours and Soft Tissue and Bone. Lyon: IARC; 2013. p. :10-1.
- Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod Pathol. 2016;29:4-13.
- [Google Scholar]
- Malignant peripheral nerve sheath tumor (MPNST) in the spine: A retrospective analysis of clinical and molecular prognostic factors. J Neurooncol. 2015;122:349-55.
- [Google Scholar]
- Nestin expression as a new marker in malignant peripheral nerve sheath tumors. Pathol Int. 2007;57:60-7.
- [Google Scholar]
- Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57:2006-21.
- [Google Scholar]
- Malignant peripheral nerve sheath tumors (MPNST) – Clinicopathological study and treatment outcome of twenty-four cases. World J Surg Oncol. 2006;4:55.
- [Google Scholar]
- Malignant tumours of peripheral nerve. In: Enzinger and Weiss's Soft Tissue Tumours (5th ed). Philadelphia: Elsevier; 2001. p. :903-4.
- [Google Scholar]
- Von Deimling A, Perry A, Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, eds. WHO Classification of Tumours of the Central Nervous System. Lyon: IARC; 2007. p. :8-9.
- Peripheral nerve sheath tumors: An electron microscopic study of 43 cases. Cancer. 1982;49:273-87.
- [Google Scholar]
- Neurofibroma (Including variants) In: Fletcher CD, Bridge JA, Hongendroorn PC, Mertens F, eds. WHO Classification of Tumours and Soft Tissue and Bone. Lyon: IARC; 2013. p. :174-6.
- [Google Scholar]
- Schwannoma (Including variants) In: Fletcher CD, Bridge JA, Hongendroorn PC, Mertens F, eds. WHO Classification of Tumours and Soft Tissue and Bone. Lyon: IARC; 2013. p. :170-2.
- [Google Scholar]
- “Ancient” schwannoma of hypopharynx: A case report with review of literature. Indian J Otolaryngol Head Neck Surg. 2011;63:60-1.
- [Google Scholar]
- Melanotic schwannoma. In: Fletcher CD, Bridge JA, Hongendroorn PC, Mertens F, eds. WHO Classification of Tumours and Soft Tissue and Bone. Lyon: IARC; 2013. p. :173.
- [Google Scholar]
- Psammomatous melanotic schwannoma as a component of Carney complex. Indian J Pathol Microbiol. 2015;58:368-70.
- [Google Scholar]
- Microcystic/reticular schwannoma of the proximal sigmoid colon: Case report with review of literature. Arch Pathol Lab Med. 2013;137:284-8.
- [Google Scholar]
- Melanotic schwannoma of thoracic spinal root mimics metastatic melanoma: A potential pitfall for misdiagnosis. Int J Clin Exp Pathol. 2015;8:8639-41.
- [Google Scholar]
- Cutaneous microcystic/reticular schwannoma: A poorly recognized entity. J Cutan Pathol. 2016;43:93-100.
- [Google Scholar]
- Primary adrenal microcystic/reticular schwannoma: Clinicopathological and immunohistochemical studies of an extremely rare case. Int J Clin Exp Pathol. 2015;8:5808-11.
- [Google Scholar]
- Peripheral nerve sheath tumours – A short series with some uncommon variants. Indian J Pathol Microbiol. 2003;46:204-6.
- [Google Scholar]
- Diagnosis of peripheral nerve sheath tumors around the pelvis. Jpn J Clin Oncol. 2004;34:405-13.
- [Google Scholar]
- Malignant peripheral nerve sheath tumour. In: Fletcher CD, Bridge JA, Hongendroorn PC, Mertens F, eds. WHO Classification of Tumours and Soft Tissue and Bone. Lyon: IARC; 2013. p. :187-9.
- [Google Scholar]
- Immunohistochemical and molecular analysis of p53, MDM2, proliferating cell nuclear antigen and Ki67 in benign and malignant peripheral nerve sheath tumours. Virchows Arch. 1995;427:19-26.
- [Google Scholar]
- Perineuroma. In: Fletcher CD, Bridge JA, Hongendroorn PC, Mertens F, eds. WHO Classification of Tumours and Soft Tissue and Bone. Lyon: IARC; 2013. p. :176-7.
- [Google Scholar]
- Granular cell tumour. In: Fletcher CD, Bridge JA, Hongendroorn PC, Mertens F, eds. WHO Classification of Tumours and Soft Tissue and Bone. Lyon: IARC; 2013. p. :178-9.
- [Google Scholar]
- Granular cell tumor of the scrotum in a child with Noonan syndrome. Pediatr Dermatol. 2008;25:341-3.
- [Google Scholar]
- Dermal nerve sheath myxoma. In: Fletcher CD, Bridge JA, Hongendroorn PC, Mertens F, eds. WHO Classification of Tumours and Soft Tissue and Bone. Lyon: IARC; 2013. p. :179-80.
- [Google Scholar]
- Solitary circumscribed neuroma. In: Fletcher CD, Bridge JA, Hongendroorn PC, Mertens F, eds. WHO Classification of Tumours and Soft Tissue and Bone. Lyon: IARC; 2013. p. :181-2.
- [Google Scholar]
- Ectopic meningioma/meningothelial hamartoma. In: Fletcher CD, Bridge JA, Hongendroorn PC, Mertens F, eds. WHO Classification of Tumours and Soft Tissue and Bone. Lyon: IARC; 2013. p. :182-3.
- [Google Scholar]
- Nasal glial heterotopias. In: Fletcher CD, Bridge JA, Hongendroorn PC, Mertens F, eds. WHO Classification of Tumours and Soft Tissue and Bone. Lyon: IARC; 2013. p. :183-4.
- [Google Scholar]
- Benign triton tumour. In: Fletcher CD, Bridge JA, Hongendroorn PC, Mertens F, eds. WHO Classification of Tumours and Soft Tissue and Bone. Lyon: IARC; 2013. p. :185.
- [Google Scholar]
- Intraneural hybrid neurofibroma/schwannoma in scalp: A case report. J Clin Diagn Res. 2015;9:ED05-6.
- [Google Scholar]
- Malignanat granular cell tumour. In: Fletcher CD, Bridge JA, Hongendroorn PC, Mertens F, eds. WHO Classification of Tumours and Soft Tissue and Bone. Lyon: IARC; 2013. p. :190.
- [Google Scholar]
- Ectomesenchymoma. In: Fletcher CD, Bridge JA, Hongendroorn PC, Mertens F, eds. WHO Classification of Tumours and Soft Tissue and Bone. Lyon: IARC; 2013. p. :191.
- [Google Scholar]
- Neurofibromatosis type 2. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, eds. WHO Classification of Tumours of the Central Nervous System. Lyon: IARC; 2007. p. :210-4.
- [Google Scholar]




