
Translate this page into:
Neuromyelitis optica and neuromyelitis optica spectrum disorder: Natural history and long-term outcome, an Indian experience
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background:
Neuromyelitis optica (NMO) has evolved from devic's classical description to a broader disease spectrum, from monophasic illness to a polyphasic illness with multiple recurrences, disease confined to optic nerve and spinal cord to now brain stem, cerebrum and even endocrinopathy due to hypothalamic involvement.
Objectives:
To report, the epidemiological characteristics, clinical presentations, recurrence rate, treatment and response to therapy in 26 patients with NMO and NMO spectrum disorder among the Indian population.
Methods:
We performed observational, retrospective analysis of our prospectively maintained data base of patients with NMO, longitudinally extensive transverse myelitis during the period of January 2003–December 2012 who satisfied the national multiple sclerosis society (NMSS) task force criteria for diagnosis of NMO and NMO spectrum disorder.
Results:
There were 26 patients (female: male, 21:5), the mean age of onset of symptom was 27 years (range 9–58, standard deviation = 12). Twenty-one patients (80%) fulfilled NMSS criteria for NMO while rest 5 patients (20%) were considered as NMO spectrum disorder. Seven patients (27%) had a monophasic illness, 19 patients (73%) had a polyphasic illness with recurrences. The Median recurrence rate was 4/patient in the polyphasic group. 13 (50%) patient were tested for aquaporin 4 antibody, 8 (61%) were positive while 5 patients (39%) were negative. All patients received intravenous methyl prednisolone, 9 patients (35%) required further treatment for acute illness in view of unresponsiveness to steroids. Thirteen patients (50%) received disease-modifying agents for recurrences. Mean duration of follow-up was 5 years. All patients had a good outcome (modified Rankin scale, <3) except one who had poor visual recovery.
Conclusion:
Neuromyelitis optica/NMO spectrum disorder is demyelinating disorder with female predominance, polyphasic course, myelitis being most common event although brain stem involvement is not uncommon with NMO antibody positivity in 60% patients, confirms the literature data.
Keywords
Aquaporin-4
longitudinally extensive transverse myelitis
neuromyelitis optica
optic neuritis

Introduction
The association between optic neuritis (ON) and spinal cord impairment was first described by Sir Clifford Albutt in 1870.[1] In 1894, Eugene Devic and his student Fernand Gault evaluated further cases and proposed the nature of the pathological process, named the syndrome neuro-mye’ lite optique or neuropticomye’ lite, and discussed a relationship with MS.[1] However, it was not until the 1990s that further clinical and histopathological studies changed the concept and place of neuromyelitis optica (NMO) within the expanding range of autoimmune disorders of the central nervous system (CNS). Now, NMO is recognized as a recurrent autoimmune CNS disorder with distinct clinical, neuroimaging and laboratory findings.[2] The cases of NMO have been reported in all continents and races, but the ethnic variations suggest, genetic factors may be important.[3456] In last decade, NMO has evolved from devic's classical description to a broader disease spectrum, from monophasic illness to a polyphasic illness with multiple recurrences, disease confined to optic nerve and spinal cord to now brain stem, cerebrum and even endocrinopathy due to hypothalamic involvement.[7] Aquaporin 4 (AQP4) antibody, an autoantibody that binds to the water channel AQP4, in combination with diagnostic criteria support the distinction of NMO from other autoimmune disorders of the CNS.[7] National multiple sclerosis society (NMSS) task force has described criteria for the diagnosis of NMO and NMO spectrum disorder with major and minor criteria.[8] Major criteria includes (i) ON in one or two eyes, (ii) transverse myelitis, clinically complete or incomplete, but associated with radiological evidence of spinal cord lesion extending over three or more spinal segments on T2-weighted magnetic resonance imaging (MRI) images and hypointensities on T1-weighted images when obtained during acute episode of myelitis, that is, longitudinally extensive transverse myelitis (LETM) and (iii) no evidence for sarcoidosis, vasculitis, clinically manifest systemic lupus erythematosus or Sjogren syndrome, or other explanation for the syndrome. All major criteria are required but may be separated by an unspecified interval. Minor criteria, from which at least one must be fulfilled, consist of (1 and/or 2): (1) Most recent brain MRI scan must be normal or may show abnormalities not fulfilling the Barkhof criteria used for McDonald diagnostic criteria including: (a) Nonspecific brain T2-signal abnormalities not satisfying the Barkhof criteria for dissemination in space used in the revised McDonald criteria, (b) lesions in the dorsal medulla, either in contiguity or not in contiguity with a spinal cord lesion, (c) hypothalamic and/or brainstem lesions, (d) linear periventricular/corpus callosum signal abnormality, but not ovoid, not extending into the parenchyma of the cerebral hemispheres in Dawson finger configuration. (2) A positive test in serum or cerebrospinal fluid (CSF) for NMO-IgG/AQP4 antibodies.[9] A monophasic form was defined by the absence of further relapses after the index events of ON and myelitis.
The mainstay of treatment for NMO is immunosuppressive therapy but due to the uncommon nature of this disease, large controlled clinical trials are difficult and treatments are based on small case series. Steroids, azathioprine, intravenous immunoglobulin (IV IgG), plasmapheresis as well as an anti-CD20 monoclonal antibody have been used in various studies.[9] If untreated, it is associated with high morbidity in the form of visual loss, quadriplegia and mortality; although there is significant improvement in outcome with early and rapid treatment as compared to previous studies.[10111213] Data regarding long-term follow-up of these patients especially from developing countries where it is still under-diagnosed and misdiagnosed, is scarce. Here we report, the epidemiological characteristics, clinical presentations, recurrence rate, treatment and response to therapy in 26 patients with NMO and NMO spectrum disorder.
Methods
The study population consisted of patients with NMO, LETM from our prospectively maintained database during the period of January 2003–December 2012 who satisfied the NMSS criteria. Patients with recurrent ON or recurrent transverse myelitis with longitudinally extensive spinal cord lesions and seropositive for AQP4 autoantibodies were classified as NMO spectrum diseases.[14] The outcome was measured using modified Rankin scale (MRS). The patient who had relapses and remission with variable degree of recovery between episodes were labeled as polyphasic illness as compared to patients with simultaneous or closely related ON and LETM (<30 days) without recurrence, that is, monophasic illness. We performed observationally, retrospective analysis of this cohort, to study the epidemiological characteristics, clinical presentations, recurrence rate, treatment and response to therapy.
Results
There were 26 patients (female: male, 21:5), the mean age of onset of symptom was 27 years (range 9–58 years, standard deviation [SD] =12) [Figure 1]. Twenty-one patients (80%) fulfilled NMSS criteria for NMO while rest 5 patients (20%) were considered as NMO spectrum disorder. Only three patients (11.5%) had a preceding history of viral illness, all in the polyphasic group. Seven patients (27%) had a monophasic illness, 19 patients (73%) had a polyphasic illness (female: male, 15:4). The median recurrence rate was 4/patient in the polyphasic group. Two patients had two relapses, five patients each had three and four relapses, two patients each had five and six relapses, seven and eight relapses in one patient each.
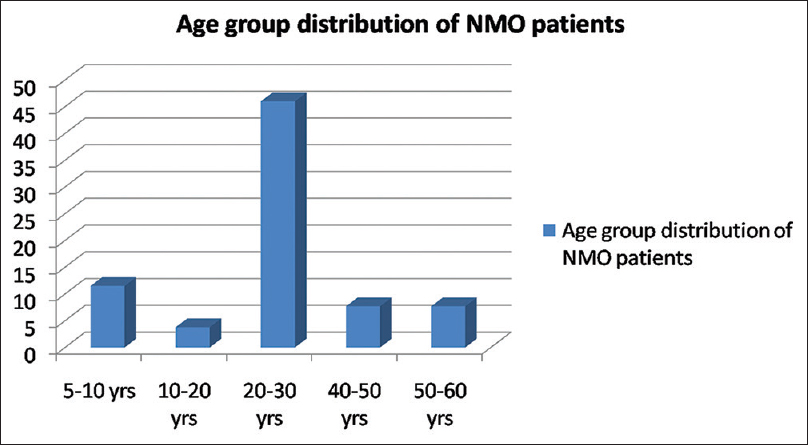
- Bar diagram showing percentage wise distribution of patients in various age groups
Among total 83 recurrent events, optic nerve alone was involved in 23%, LETM in 56.62%, both (ON and LETM) in 16.9% while brain stem features were present in 3.6% of patients in recurrence group [Figure 2]. One patient presented with recurrent vomiting before the development of myelitis[15] [Figure 3]. Eight patients (33%) showed CSF pleocytosis (predominantly lymphocytes). Maximum cell count in CSF was 75/mm3 (range 2–75, mean 11, SD = 16). CSF protein ranged from 18 mg% to 70 mg%. Thirteen (50%) patient were tested for serum AQP4 antibody in the serum, out of which 8 (61%) were positive while 5 patients (39%) were negative. All patients received intravenous methyl prednisolone, 9 patients (35%) required further treatment for acute illness in view of unresponsiveness to the steroids. Of these, six patients (27%) received plasma-pheresis while 2 patients (12%) received IV IgG. One patient needed rescue plasma exchange when symptom worsening was noticed despite IV IgG treatment [Table 1].
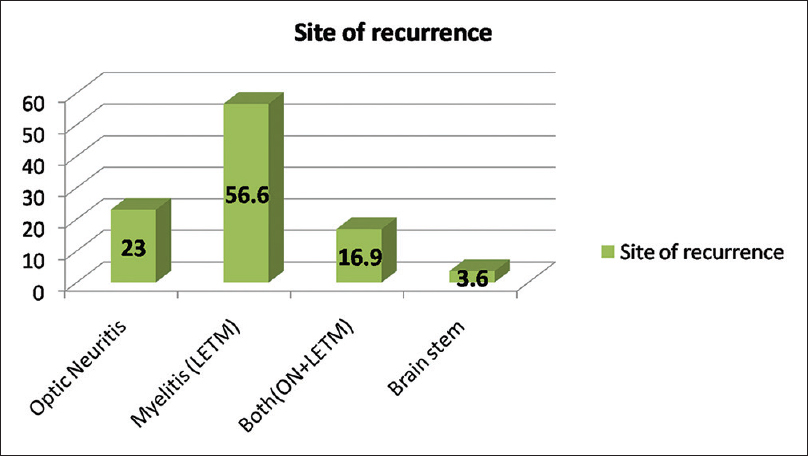
- Bar diagram showing percentage wise distribution of site of recurrence
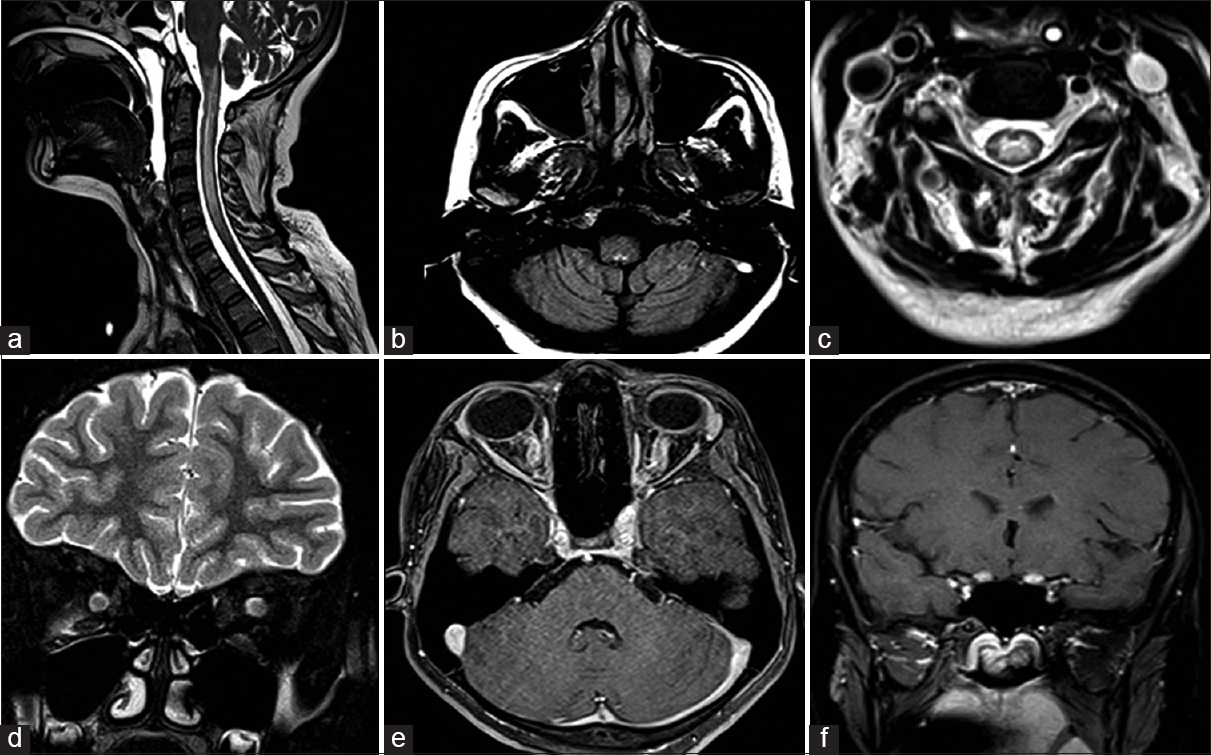
- (a) Sagittal T2-weighted cervical spine and brain stem, depicting patchy hyperintensities in cervico-medullary junction and segmental involvement of cervical spinal cord. (b) Axial fluid attenuation inversion recovery (FLAIR) image at the level of posterior fossa, demonstrating hyperintensity in the dorsal medullary region. (c) Axial FLAIR image at cervical level showing central cord hyperintensity. (d) Coronal T2-weighted image showing bulky optic nerves bilaterally. (e) Axial VIBE sequence showing enhancement of bilateral optic nerve. (f) T1 Postcontrast image showing enhancement extending up to prechiasmatic region
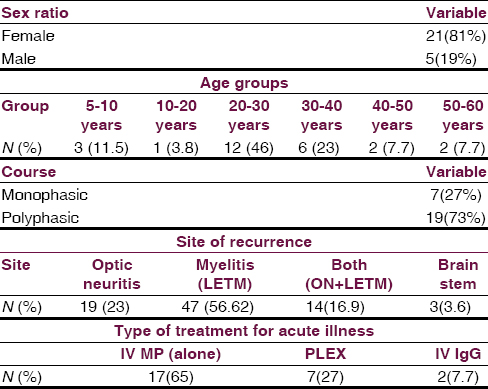
Thirteen patients (50%) received disease-modifying agents for recurrences. (Azathioprine in 9, methotrexate in 1, mitoxantrone in 1 while monthly IV MP in 2). The mean duration of follow-up was 5 years. Patients who received monthly IV MP and Mitoxantrone were switched to azathioprine during recurrent illness when a correct diagnosis of NMO was made instead of previous diagnosis of MS. All patients had good outcome (MRS <3) except one who had poor visual recovery and no mortality was observed.
There was no difference in clinical presentation, CSF characteristics, AQP4 antibody positivity or long-term outcome of illness in the patients whose acute illness responded to IV MP alone or required other immunotherapy (IV IgG or plasmapheresis).
Discussion
Neuromyelitis optica makes up a substantial proportion of inflammatory demyelinating disorders of the CNS in noncaucasian populations such as Afro-Brazilians (15%), East Asians (up to 48%) and Indians (9%).[16171819] Previous studies has shown that in monophasic NMO men and women are affected equally but in the more frequent recurrent disease course (80–90%), women (ratio 5–10:1) are overrepresented.[156] The similar results were obtained in our study also with female to male ratio of 4:1, while in a previous study from India male predominance was noted, although, it could be due to bias towards male patients for admission and treatment.[12] The age of onset in NMO ranges from childhood to late adulthood with the decrease in the incidence after the fifth decade; the median age of onset is in the late 30's.[202122] The mean age of onset in our study was 27 years, comparable to western cohorts.[202122] An antecedent viral illness was reported in 30% of patients with monophasic and 23% of patients with recurrent NMO,[6] whereas 10% patients with polyphasic NMO. In our study cohort only 3 patients (11.5%) had preceding viral illness in the polyphasic group. In most of the series of NMO, recurrent course (80–90%) is common, while monophasic (10–20%) and primary or secondary progressive courses are rare.[161023] Similarly, the polyphasic course was noted in 73% of our patients. Intractable nausea and hiccough as a presentation of NMO due to inflammatory lesions in the dorsal medulla involving area prostema has been described in literature.[15]
Previous studies have shown that CSF abnormalities are detected in most patients with NMO and concern cell count, protein level and oligoclonal bands. CSF pleocytosis, usually consisting of monocytes and lymphocytes, is present in 14–79% of patients. CSF pleocytosis can include or be dominated by neutrophils, also eosinophils may be found. CSF cell count is >50 cells/cuml in 13–35% of patients and in a few cases up to 1000 cells/cuml.[624] In our study, CSF pleocytosis was noted in 8/26 patients (33%), predominantly lymphocytes with the maximum cell count in CSF was 75/mm3. As NMO-IgG is 91–100% specific and 70–93% sensitive in NMO, has become a reliable tool to distinguish NMO spectrum of disorders from other demyelinating illnesses.[7] In our study 13 (50%) patient were tested for AQP4 antibody, out of which 8 (61%) were positive. In a previous study from India[12] only 1 out of 8 cases (12.5%) of NMO had shown NMO antibody positivity, and the author even concluded that in Indian population, the NMO antibody positivity is less in comparison to western world, but our results are similar to western cohort.
In polyphasic group, 83 recurrent events occurred in 19 patients, in which optic nerve alone was involved in 23%, LETM in 56.62%, both (ON and LETM) in 16.9% and brain stem in 3.6% of patients, similar to previous studies.[161022] The patient Median recurrence rate was 4/patient with a variable interval in between. Our 65% patients responded to IV MP, 27% to plasmapheresis and 7.7% to IV IgG, all had excellent recovery.
As for as outcome is concerned, in the study by Wingerchuk and Weinshenker[10] 32% of patients with recurrent NMO died (median follow-up time 60.2 months) while no death occurred in patients with monophasic NMO showing probable bias towards more severe and complicated cases. Similarly, there were 24/96 (25%) deaths in the French West Indies cohort,[11] while mortality in a cohort of recurrent NMO from Brazil was even higher (50%).[18] In contrast, our study had shown a good outcome (m-RS <3) with no direct NMO related mortality, morbidity in the form of a single patient with poor visual recovery was noted. Similar types of results were noted from India by Pandit[12] There were similar reports of favorable outcome from other centers in India. Pradhan and Mishra[13] had described 6 patients (3 male and 3 female) with clinical and radiological features consistent with relapsing NMO who recovered well after recurrent attacks of visual loss and myelitis, remaining ambulant 2–10 years after onset of disease. The imaging characteristics of patients with early or late onset disease and their responsiveness to steroid did not differ. In addition to being retrospective, our study has other limitations. First, we have small number of cases, second included only patients who were hospitalized resulting in selection bias.
Conclusion
Neuromyelitis optica/NMO spectrum disorder is demyelinating disorder with female predominance, polyphasic course, myelitis being most common event although brain stem involvement is also seen with NMO antibody positivity in 60% patients, confirms the literature data.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- Clinical, CSF, and MRI findings in Devic's neuromyelitis optica. J Neurol Neurosurg Psychiatry. 1996;60:382-7.
- [Google Scholar]
- NMO-IgG predicts the outcome of recurrent optic neuritis. Neurology. 2008;70:2197-200.
- [Google Scholar]
- Neuromyelitis optica in Brazil: A study on clinical and prognostic factors. Mult Scler. 2009;15:613-9.
- [Google Scholar]
- Characterisation of the spectrum of demyelinating disease in Western Australia. J Neurol Neurosurg Psychiatry. 2008;79:1022-6.
- [Google Scholar]
- Characteristics of Devic's disease (neuromyelitis optica) in Mexico. J Neurol. 2008;255:710-5.
- [Google Scholar]
- The clinical course of neuromyelitis optica (Devic's syndrome) Neurology. 1999;53:1107-14.
- [Google Scholar]
- Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: A study on antibody titre. Brain. 2007;130:1235-43.
- [Google Scholar]
- Differential diagnosis of suspected multiple sclerosis: A consensus approach. Mult Scler. 2008;14:1157-74.
- [Google Scholar]
- An open label study of the effects of rituximab in neuromyelitis optica. Neurology. 2005;64:1270-2.
- [Google Scholar]
- Neuromyelitis optica: Clinical predictors of a relapsing course and survival. Neurology. 2003;60:848-53.
- [Google Scholar]
- Relapsing neuromyelitis optica: Long term history and clinical predictors of death. J Neurol Neurosurg Psychiatry. 2009;80:1162-4.
- [Google Scholar]
- Neuromyelitis optica antibody (NMO-IgG) status in Indian patients with multiple sclerosis and allied demyelinating disorders. Neurol Asia. 2008;13:175-8.
- [Google Scholar]
- A central demyelinating disease with atypical features. Mult Scler. 2004;10:308-15.
- [Google Scholar]
- Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol. 2006;59:566-9.
- [Google Scholar]
- Intractable nausea and vomiting as presenting manifestation of neuromyelitis optica. Ann Indian Acad Neurol. 2013;16:400-1.
- [Google Scholar]
- A retrospective review of patients with clinically definite multiple sclerosis. Ann Acad Med Singapore. 1998;27:204-9.
- [Google Scholar]
- Optic neuromyelitis syndrome in Brazilian patients. J Neurol Neurosurg Psychiatry. 2002;73:429-35.
- [Google Scholar]
- Neuromyelitis optica-IgG in childhood inflammatory demyelinating CNS disorders. Neurology. 2008;70:344-52.
- [Google Scholar]
- A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology. 2007;68:603-5.
- [Google Scholar]
- Devic's neuromyelitis optica: Clinical, laboratory, MRI and outcome profile. J Neurol Sci. 2002;197:57-61.
- [Google Scholar]




