
Translate this page into:
Myxoid chondrosarcoma of sphenoid bone
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
The myxoid variant of chondrosarcoma is usually seen in soft tissues where it is known as chordoid sarcoma or parachordoma. Rarely, it involves bone and when it does, cranial bones are the preferred location. This tumor is frequently amalgamated with the chondroid variant of chordoma, especially when the lesion occurs in the sphenoid bone/spheno-occipital region, because of their similar clinical presentations, anatomical locations, radiological findings, and mistaken histopathological features. It is essential to distinguish myxoid chondrosarcoma from the chondroid variant of chordoma, because of the different treatment protocol and prognostic importance. We present such a location-based diagnostic dilemma, solved successfully with ancillary immunohistochemistry.
Keywords
Chondrosarcoma
chordoma
immunohistochemistry
sphenoid bone

Introduction
Chondrosarcoma and chordoma of the skull base are rare tumors. The combined incidence is reported to be 0.03 per 100,000 persons in the United States.[1] The myxoid variant of chondrosarcoma of bony origin is an uncommon entity and its location in the skull is rarer still with incidence as low as 0.16%, including those arising from its coverings, supporting tissues, and cranial bones.[2] This tumor occurs mainly in the spheno-occipital region. Because of the similarity to chordoma, the synonym for it is chordoid sarcoma or parachordoma, and indeed, it is misdiagnosed frequently as such. It is essential to distinguish myxoid chondrosarcoma from chordoma and especially its chondroid variant, as it has a better prognosis when compared to the latter.
Case Report
A 48-year-old female presented with headache and sixth cranial nerve palsy since six months and vomiting since 20 days. There was no history of hypertension, diabetes mellitus, or asthma. No source of any oral or respiratory infection was noted. She had no significant medical or surgical history in the past. Computed tomography scan (CT scan) showed a lobular lesion measuring 3.9×3.0 cm with calcific specks in the medial temporal region involving the left greater wing and body of the sphenoid bone along with the petrous part of the temporal bone. Magnetic resonance imaging (MRI) showed a T2 hyperintense lesion involving the clivus, petrous temporal bone, and sphenoid bone on the left side. This lesion was hypointense on T1- weighted image [Figure 1a] and enhancing heterogeneously in postcontrast (gadolinium) study [Figure 1b]. As the lesion was in midline and contained calcified matrix, a provisional diagnosis of chordoma was made. Perioperatively, the tumor was noted to be of extradural location, and was soft and suckable. The excised tissue comprised multiple irregular, glistening, soft lesional fragments of 0.4 to 0.8 cm with focal gritty areas of calcification.
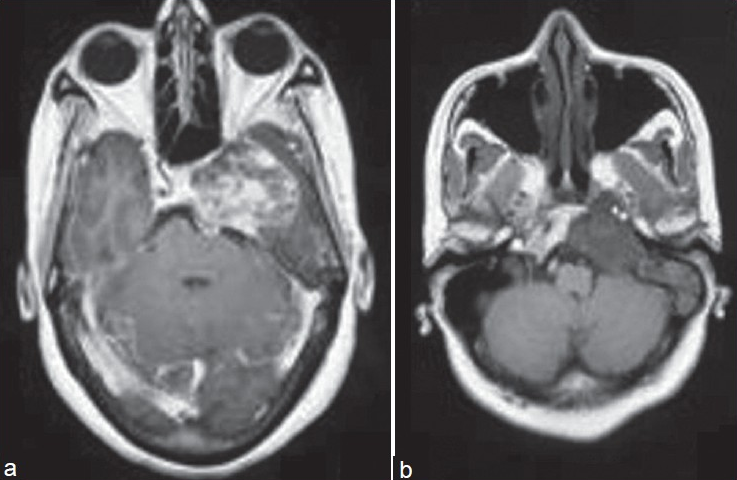
- (a) T1-weighted axial image: Lesion is hypointense on T1-weighted image (b) Postcontrast T1-weighted axial image: Shows heterogeneous enhancement of the lesion
Pathological findings
Fragmented tissue samples from the mass were fixed in 10% formalin, routinely processed for paraffin embedding, and stained with hematoxylin and eosin. The histological sections revealed the lobular lesional component [Figure 2] comprising large cells with round to mildly pleomorphic vesicular nuclei and abundant vacuolated cytoplasm lying on a mucomyxoid background [Figure 3]. These were associated with conspicuous cartilaginous areas having the presence of uni to multinucleated lacunae, foci of dystrophic calcification and intervening fibrocollagenous tissue with proliferating congested capillaries, separating the lobules. A histological diagnosis of myxoid variant of chondrosarcoma was made. To rule out the clinical and radiological suspicion of chordoma and to affirm the hematoxylin and eosin diagnosis, the help of immunohistochemistry was sought. The immunohistochemical positivity of the tumor component for the S100 protein and vimentin [Figure 4] and negativity for the epithelial membrane antigen (EMA) and cytokeratin (CK)favored the diagnosis of myxoid variant of chondrosarcoma over chondroid chordoma.

- Lobular arrangement of tumor cells separated by fibrous septae [ hematoxylin and eosin × 100]

- Large bubbly-looking tumor cells with stellate-shaped nucleus and abundant vacuolated cytoplasm (physaliphorous-like cells) [hematoxylin and eosin × 400]

- Cytoplasmic positivity for vimentin [immunohistochemistry × 400]
Discussion
Extraskeletal myxoid chondrosarcoma (EMC) was first described in 1953 by Stout and Verner,[3] who reported a small series of morphologically similar appearing cases, all of which arose in the extremities of adults. However, in 1972, Enzinger and Shiraki,[4] in a series of 34 cases, first established EMC or chordoid sarcoma as a distinct clinicopathologic entity. Despite numerous additional case reports and series of extraskeletal chordoid sarcoma, only a few case reports of its skeletal counterpart, SMC (SMC: skeletal myxoid chondrosarcoma) are found in the literature.
Are SMC and EMC the same entity arising in two different locations, or are they two separate entities? In a systematic attempt to answer the question, Antonescu et al.[5] performed a comparative clinicopathologic, ultrastructural, and molecular study of SMC and EMC. The gender distribution was identical in both groups and the mean age was slightly higher for EMC patients [55 vs 45 years]. Electron microscopy revealed intracisternal microtubules and prominent mitochondria in EMC cases and only inconspicuous organelles in SMC. Molecular analysis for the EWS-CHN fusion RNA resulting from the t (9;22) was detected in EMC cases only. The conclusion drawn was that although similar light microscopic features are noted in EMC and SMC, fundamental differences are present at the ultrastructural and molecular levels, suggesting that EMC and SMC represent two distinct entities in the chondrosarcoma family of tumors.
Chordoma is a slow-growing malignant neoplasm arising from ectodermally derived notochordal remnants and accounts for less than 5% of primary bone tumors. Morphologically, chordoma is composed of large cohesive cells with abundant, bubbly eosinophilic cytoplasm, known as ‘physaliphorous cells’. Hyaline cartilage is rarely observed except in the variant located in spheno-occipital region, the so-called chondroid chordoma. Based solely on microscopic features, distinction between chordoid sarcoma and chordoma may be difficult as the vacuolated areas of chordoid sarcoma may closely resemble those observed in chordoma. However, diagnostic physaliphorous cells are absent in chordoid sarcoma in contrast to chordoma, and cytoplasmic vacuolation tends to be focal rather than diffuse. Further, although both the lesions are multilobated, this feature tends to be more distinct in chordoid sarcoma.
In 1973, Heffelfinger et al.,[6] for the first time described a chordoma with cartilaginous differentiation and named it the chondroid variant of chordoma which later on gained support from the immunohistochemical study by Rosenberg et al.,[7] who demonstrated positivity for epithelial and mesenchymal markers (i.e., EMA, CK, S100 protein, and vimentin) in both chordoma and its chondroid variant while differentiating the myxoid variant of chondrosarcoma which displayed positivity only for the mesenchymal markers (i.e., S100 protein and vimentin).
For diagnosing cases without classical histology, that is, myxoid chondrosarcoma with atypical morphological features, apart from immunohistochemistical study, FISH (FISH: fluorescent in situ hybridization) analysis using EWSR1 and NR4A3 probes has been proposed by Noguchi et al.[8] In their study comprising 18 cases of EMC, the positive results of 72% for EWSR1 and 61% for NR4A3 gene rearrangements led to the conclusion that FISH analysis of formalin-fixed, paraffin-embedded specimens for the said new probes is useful, convenient, and may provide an ancillary method for diagnosis, especially in difficult cases.
Remnants of primitive notochord at the spheno-occipital synchondrosis gives rise to skull chordomas whereas chondrosarcomas take origin from the embryonic rest of the cartilaginous matrix of the cranium or primitive mesenchymal cells. Chondrosarcomas usually begin laterally (i.e., from the spheno-occiput region) and grow into the midline to involve the clivus, whereas chordomas behave vice versa.[2]
In an immunohistochemical study to predict clinical outcome, Heyse et al.[9] compared three isoforms of CD44 and came up with the conclusion that overexpression of CD44s correlated significantly with metastatic potential and poorer survival of patients with chondrosarcoma, which may serve as a prognostic marker independent of grading and other covariates.
Nguyen et al.[10] proposed adjuvant radiation therapy after maximal surgical resection of the tumor. After comparing with other forms of radiation therapy, they advocated proton beam therapy as a better tool used to increase the dose delivered to the tumor while elegantly sparing dosing to adjacent critical normal structures.
To conclude, although the myxoid variant of chondrosarcoma and chondroid chordoma are similar in several aspects, they differ in their origin and clinical outcome. Skull base myxoid chondrosarcoma carries a markedly more favorable outcome with minimal recurrence and excellent long-term control, whereas chordomas including its chondroid variant demonstrate an aggressive clinical course and have a uniformly poor outcome after disease recurrence. Radiotherapy is usually recommended following surgery in chordoma, whereas resection of the tumor suffices in the myxoid variant of chondrosarcoma. Our patient underwent surgical excision of the tumor without any radiotherapy and is doing well in the postoperative one-year period. Although a few cases of myxoid chondrosarcoma of the sphenoid bone have been described in literature, our case adds to the existing literature emphasizing the importance of immunohistochemistry in arriving at a final diagnosis.
Acknowledgment
The authors wish to thank the senior technical staff, Mrs. Ushanandini and Mr. Ramanna for the immunohistochemical stains.
Source of Support: Nil
Conflict of Interest: None declared
References
- CBTRUS 2005. Central Brain Tumor Registry of the United States 2005-2006. Statistical Report 1998-2002 Years Data Collected
- [Google Scholar]
- Chordoma and chondrosarcoma: Similar, but quite different, skull base tumors. Cancer. 2007;110:2457-67.
- [Google Scholar]
- Extraskeletal myxoid chondrosarcoma: An analysis of 34 cases. Hum Pathol. 1972;3:421-35.
- [Google Scholar]
- Skeletal and extraskeletal myxoid chondrosarcoma: A comparative clinicopathologic, ultrastructural, and molecular study. Cancer. 1998;83:1504-21.
- [Google Scholar]
- Chondroid chordoma - a variant of chordoma.A morphologic and immunohistochemistry study. Am J Clin Pathol. 1994;101:36-41.
- [Google Scholar]
- Fluorescence in situ hybridization analysis of extraskeletal myxoid chondrosarcomas using EWSR1 and NR4A3 probes. Hum Pathol. 2010;41:336-42.
- [Google Scholar]
- CD44: Survival and metastasis in chondrosarcoma. Osteoarthritis Cartilage. 2010;18:849-56.
- [Google Scholar]
- Emerging role of proton beam radiation therapy for chordoma and chondrosarcoma of the skull base. Curr Oncol Rep. 2008;10:338-43.
- [Google Scholar]




