
Translate this page into:
Meckel-Gruber syndrome: Report of two cases
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Meckel-Gruber syndrome (MKS) is an autosomal recessive disorder, characterized by a combination of renal cysts and variably associated with features including developmental anomalies of the central nervous system (typically encephalocele), hepatic ductal dysplasia, cysts, and polydactyly. It is a rare syndrome with highest incidence in Gujarati Indians and Finnish population. We report two such cases of MKS in non-Gujarati Indian which were diagnosed by neonatal autopsy.
Keywords
Meckel-Gruber syndrome
occipital encephalocele
polycystic kidney
polydactyly

Introduction
Meckel-Gruber syndrome (MKS) is an autosomal recessive, lethal syndrome characterized by a wide variety of systemic malformations. The most consistent are occipital encephalocele, multicystic dysplasia of kidney, cystic and fibrotic changes of liver, and polydactyly.[1] The worldwide incidence of MKS varies from 1 in 13,250 to 1,40,000 live births.[2] Antenatal ultrasound examination (USG) can identify the major features, but a neonatal autopsy is needed to document complete anomalies and for syndrome diagnosis. We report two such cases which were diagnosed by neonatal autopsy.
Case Reports
Case 1
A 22-year-old primigravida presented with 19 weeks of gestation for routine antenatal examination. There was history of second-degree consanguineous marriage. Past and family histories were noncontributory. She was not on any teratogenic drugs. Routine antenatal scan done showed features of anencephaly. With the consent of parents, termination of the pregnancy was undertaken. The abortus was sent to histopathological examination. At autopsy, it was a male fetus weighing 150g; on external examination, there was a defect in the occipital bone with microcephaly and encephalocele through the posterior fontanelle. There was micro-ophthalmia and oral cavity showed cleft lip with cleft palate. All the four limbs showed presence of post-axial polydactyly. Left club foot was also seen. On dissection, the herniation of the brain was confirmed, cerebellar hypoplasia was present. Liver showed biliary duct hyperplasia and fibrosis. Kidneys were grossly normal and microscopic examination showed multiple minute cysts of different size, lined by flattened epithelium [Figure 1]. The lungs were hypoplastic. Other visceral organs were normal. Based on the above features, pathological diagnosis of MKS was made.
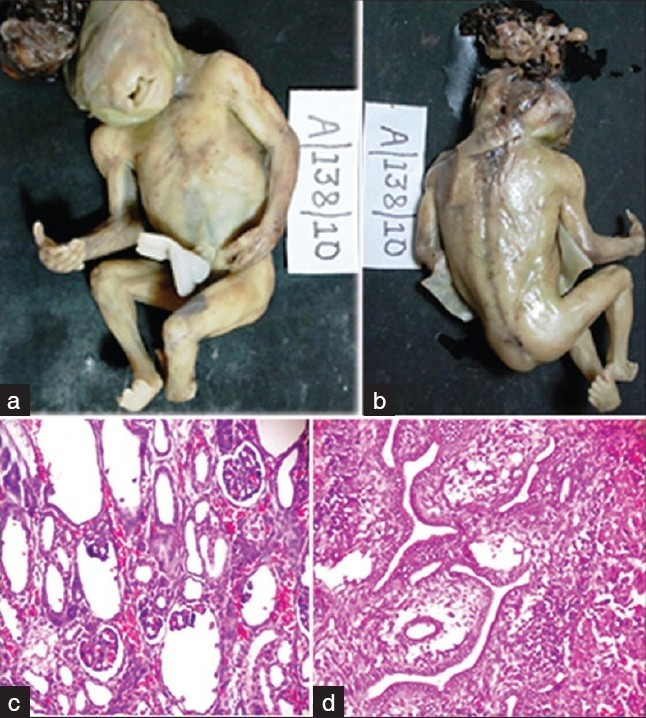
- (a) Ventral surface of fetus showing encephalocele, cleft lip, and polydactyly. (b) Dorsal surface showing defect in occipital bone, encephalocele arising from posterior fontanelles, left club foot, and polydactyly. (c) Microphotograph of the kidney showing minute multiple cysts lined by flattened epithelium (H and E, ×100). (d) Microphotograph of liver showing biliary hyperplasia and portal fibrosis (H and E,×100)
Case 2
A 24-year-old female (G4, P3, L2) presented with 18 weeks 4 days of gestation for routine antenatal examination. There was no history of consanguineous marriage. Her first male child died at 6 years, the cause was not known; other two female children are healthy. She was not on any teratogenic drugs. Routine antenatal scan done showed features of anencephaly with multicystic kidneys and the lower limb was not visualized at USG. With the consent of the parents, termination of the pregnancy was undertaken. The abortus was sent to histopathological examination. At autopsy, it was a male fetus weighing 150g; on external examination, there was microcephaly with encephalocele through the posterior fontanelle. The abdomen was distended and all the four limbs showed presence of post-axial polydactyly. On dissection, the herniation of the brain was confirmed, kidneys showed cysts of multiple sizes. On microscopic examination, the cysts were of different sizes and lined by flattened epithelium [Figure 2]. Other visceral organs were normal. Based on the presence of classical features, a diagnosis of MKS was suggested. Both of our patients’ parents did not give consent for genetic analysis.

- (a) Ventral surface of fetus showing distended abdomen and polydactyly of all four limbs. (b) Dorsal surface showing encephalocele arising from posterior fontanelle and polydactyly. (c) Gross photograph of cut surface of kidney showing multiple cysts. (d) Microphotograph of the kidney showing multiple cysts lined by flattened epithelium (H and E, ×100)
Discussion
MKS was first described by Johann Friedrich Meckel in 1822 in two siblings who died of identical malformations of occipital encephalocele, polycystic kidneys, and polydactyly. George B Gruber, in 1934, reported many familial cases with similar features and coined the term “dysencephalia splanchnocystica.”[3] In 2006, Opitz et al.[4] gave the detailed review of developmental pathology of Meckel syndrome.
The incidence of this rare syndrome is 1 per 1 300 live births in Gujarati Indian families,[5] 1 per 3 000 in Belgium, and 1 in 9 000 in Finland. The disease affects all races with males and females being equally affected. Once diagnosed, the chances of MKS in subsequent pregnancy are 1 in 4 (25%). The diagnostic criteria for MKS is presence of at least two of the three classic features like cystic renal dysplasia, occipital encephalocele, and polydactyly, which are observed in 100%, 90%, and 83.3%, respectively.[1–3] As observed in our cases, our case number 1 showed minute cyst in the kidney on microscopic examination, indicating the need of neonatal autopsy in syndrome diagnosis. The anomalies observed in MKS are listed in Table 1.[6]

MKS is highly heterogenous syndrome; six different loci have been identified. MKS 1 was mapped to chromosomes 17q21-q24 in Finnish population. The novel gene FLJ 20345/MKS1 encodes a protein MKS1, which is having 559 amino acids. The tissue expression of this gene was reported mainly in brain, liver, kidney, and cartilage of the developing digits. The second MKS locus, MKS2 mapped to chromosome 11q 13, the novel gene is yet to be identified. This locus is commonly observed in North Africa and Middle East. MKS 3 was mapped to 8q24, the novel gene MKS3/TMEM 67 was identified from MKS cases from Pakistan and Oman. The predicted protein Meckelin comprises of 995 amino acids in human beings. MKS3 is having expression in adrenal gland, brain, kidney, lung, and spinal cord explaining the rarity of polydactyly in MKS 3.[7]
Consugar et al.[8] screened the coding regions of MKS 1 and MKS 3 in 17 non-consanguineous US and Netherland families, observed both the mutations in ten families, five MKS 1 and five MKS 3. MKS1-Fin major mutations accounted for the maximum of MKS 1 mutations. They have identified 7 novel mutations in MKS 3 and concluded that MKS 1 and MKS2 accounted for the majority MKS in non-consanguineous populations of European origin. A molecular study by Paarvola et al.[1] on five families diagnosed as MKS, using four polymorphic DNA markers, showed genetic heterogeneity of MKS as there was failure of cosegregation of in three of four families. They also suggested that cerebro-renal-digital syndrome is highly heterogeneous group of diseases; hence, the diagnosis should be confirmed by autopsy and genetic studies. Molecular studies of this kind are needed in Indians Gujarati and non-Gujarati families to document the novel mutations.
The spectrum of central nervous system malformations observed in MKS ranges from total craniorachischisis at the most severe end to a partial defect of the corpus callosum at the mildest end of the spectrum. Neuropathological studies have shown that prosencephalic dysgenesis with absence of the olfactory bulbs and tract is frequent, as well as defects in midline formation sometimes associated with absence of the lateral ventricles. The occipital encephalocele usually comes through an apical defect of the occipital bone or enlarged posterior fontanelle. Often a second, smaller defect of the basal occipital bone is found with or without another meningocele. Anomalies of the rhombic roof with a large supracerebellar cyst including the fourth ventricle and complete agenesis of the cerebellar vermis have been reported. The different degrees of dysgenesis of posterior fossa structures have been described as variants of Dandy-Walker malformation.[9]
Moerman et al.[10] reported a family with two affected sibs with typical MKS including occipital encephalocele, and the other with cerebellar Dandy Walker cyst, absent vermis, arhinencephaly dilatation of the ventricles, and changes in the kidneys and liver typical of MKS. They included another similar pair of sibs and considered cerebellar Dandy-Walker malformation as one component of MKS.
MKS has to be differentiated from trisomy 13 (holoprosencephaly, cleft lip/palate, congenital heart diseases, and polydactyly), trisomy 18 (Choroid plexus cyst, congenital heart/kidney disease, rocker bottom feet, and polydactyly), Joubert Syndrome (hypoplasia/dysplasia of vermis, facial abnormalities, cystic renal diseases, polydactyly, and cleft palate), Bardet-Biedl syndrome (vision loss, mental retardation, renal diseases, polydactyly, and cleft palate), and Smith-Lemli-Opitz syndrome (Microcephaly, Mental retardations, and polydactyly). The complexity of the differential diagnosis underlines the need for molecular testing for this syndrome.[23811]
MKS can be diagnosed by USG done at 11 to 14 weeks of gestational age and by estimation of alpha fetoprotein in the maternal serum. Sometimes, alpha fetoprotein level is not elevated when the encephalocele contain a closed sac.[23] When available, neonatal autopsy and genetic study are gold standard for diagnosis.[2]
MKS is a lethal syndrome, generally results in intrauterine death or neonatal death within few hours of life. The longest survival recorded in the literature is 4 months.[3]
Conclusion
MKS is a rare neural tube defect associated with wide variety of malformations. Neonatal autopsy and genetic studies are gold standard for the diagnosis and to document the anomalies. When available, chromosomal analysis is done to confirm the diagnosis. USG and maternal serum alpha fetoprotein level estimation can be used to know the recurrence in subsequent pregnancy.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- Meckel-Gruber syndrome: pathologic manifestations, minimal diagnostic criteria and differential diagnosis. Arch Pathol Lab Med. 2006;130:1236-8.
- [Google Scholar]
- Meckel-gruber syndrome Associated with CNS malformations – A case report. Int J Pharm Biosci. 2011;2:B484-91.
- [Google Scholar]
- High incidence of Meckel's syndrome in Gujarati Indians. J Med Genet. 1985;22:301-4.
- [Google Scholar]
- Antenatal Ultrasound diagnosis of meckel-gruber syndrome. Indian J Radiol Imaging. 2001;11:199-201.
- [Google Scholar]
- The Meckel–Gruber syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum Mol Genet. 2007;16:173-86.
- [Google Scholar]
- Molecular diagnostics of Meckel–Gruber syndrome highlights phenotypic difference between MKS1 and MKS3. Hum Genet. 2007;121:591-9.
- [Google Scholar]
- The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39:875-81.
- [Google Scholar]




